Fig. 3.1
Immune response in SCI (immune system–brain interaction): (A) following initial injury, neutrophils and macrophages (monocytes) invade the lesion by activating resident microglia; (B) then, T and B cells engaged in adaptive immunity also infiltrate; (C) finally, activated astrocytes and fibroblasts further delineate the lesion to form glial and fibrotic scars. Each cell component has been shown to have a detrimental (red arrows) or supportive effect (blue arrows) on neural tissue and regrowth of axons during the healing process (see the text for details)
Although the inflammatory responses are somewhat different among the species (human, rats, and mice) and among strains within the same species (Fleming et al. 2006; Kigerl et al. 2006; Popovich et al. 1997), neutrophils are the first cells to invade the lesion, starting within the first few hours or days (Schnell et al. 1999a). Monocytes of hematopoietic origin then infiltrate within a week after SCI, with concomitant activation of resident microglial cells (Donnelly and Popovich 2008). Thereafter, the entry of lymphocytes (T and B cells) is observed in the lesion within a period of weeks to months (Fleming et al. 2006; Kigerl et al. 2006; Sroga et al. 2003). Injury signals such as adenosine triphosphate (ATP), danger-associated molecular patterns (DAMPs: heat shock proteins, HMGB1, and peroxiredoxins) and cytokines (IL-1β and TNF-α) emanate from CNS resident cells to recruit and activate circulating leukocytes (de Rivero Vaccari et al. 2012; Shichita et al. 2012). Since these cells often stay in the lesion for months, SCI could be perceived as a chronic inflammatory disease.
Other components such as glial cells (astrocytes) and fibroblasts are further involved in the repair process to create scar tissue (Silver and Miller 2004), following the entry of leukocytes derived from hematopoietic cells. Astrocytes are activated; they proliferate, and migrate or extend their processes to surround the lesion and create a glial scar. This separates the inflammatory and intact regions and helps limit the expansion of inflammation, reconstruct the BBB, and form scar tissue within 1–2 weeks (Okada et al. 2006; Wanner et al. 2013). In the core of the lesion, fibroblasts eventually accumulate and create a fibrotic scar, to fill the necrotic gap between intact neural tissues (Herrmann et al. 2010; Zukor et al. 2013).
As overviewed here, multiple players are involved in the healing process. The following sections summarize how each cellular player influences neighboring neural tissue and the recovery process.
3.2.2 Neutrophils
Neutrophils are the first responders to inflammation that occurs as a result of infections and injuries (Wright et al. 2010) (Fig. 3.1A). In fact, they start appearing in the acute phase, 3–24 h after SCI, and their numbers peak at around day 3 in mice and at 8 h in rats (Kigerl et al. 2006; Mawhinney et al. 2012; Stirling and Yong 2008; Taoka et al. 1997b). In most species, neutrophils have disappeared within the first week (Donnelly and Popovich 2008), but they persist for several months in mice (Kigerl et al. 2006; Mawhinney et al. 2012). They generally have bactericidal properties for engaging in the defense of host tissue and function via phagocytosis and the release of anti-bacterial granules into extracellular space. These granules contain reactive oxygen species (ROS), lysosomal enzymes (myeloperoxidase MPO, elastase, MMP-9), and pro-inflammatory cytokines (Wright et al. 2010). Hence, it is conceivable that this defensive strategy can simultaneously damage neighboring healthy bystander tissue as well. Indeed, it is suggested that neutrophils damage neural tissue after SCI. Inhibiting the invasion of neutrophils by leukocyte depletion, treatment with prostaglandin E2, anti-P-selectin, anti-ICAM1, or anti-CD11d antibodies to inhibit endothelial cell–neutrophil interaction, or iloprost (a synthetic analog of prostacyclin), and knockout of MMP-9 are all correlated with reduced entry of neutrophils and enhanced recovery of motor functions and histopathological events (Gris et al. 2004; Hamada et al. 1996; Naruo et al. 2003; Noble et al. 2002; Taoka et al. 1997a, b). Inhibiting the release of these factors using elastase inhibitors, and knockout of MPO, are also correlated with reduced tissue damage and recovery (Kubota et al. 2012; Taoka et al. 1998). Each modulation that reduces damage by neutrophils is well correlated with decreases in cell death, BBB permeability, production of enzymes, ROS, and pro-inflammatory cytokines, suggesting a destructive role for neutrophils. However, the actual role of neutrophils should still be carefully evaluated, since in most cases these experiments do not specifically target neutrophils. For example, ICAM-1 and P-selectin knockout (KO) mice show better recovery and tissue sparing than antibody treatments, but they also reduce macrophage/microglial invasion (Farooque et al. 1999, 2001). P2X4 knockout and anti-CD11d antibody treatments also reduce the number of neutrophils concomitantly with monocytes/macrophages and improve behavioral recovery (de Rivero Vaccari et al. 2012; Gris et al. 2004).
In contrast to their detrimental role, recent reports demonstrate that the selective depletion of neutrophils using anti-Ly6G antibody alone has no effect on recovery, whereas depletion of both neutrophils and macrophages improves recovery (Lee et al. 2011). In another study, there was a conflicting finding that depletion of neutrophils using anti-Ly6G antibody worsens pathological processes and recovery (Stirling et al. 2009). Although the reasons for these controversies are yet unknown, the timing of treatment and the specificity of the antibody to neutrophils should be further examined.
From the reports described above, it can be assumed that neutrophils are required for the initial repair process, but the excessive responses frequently seen in SCI are detrimental to tissue sparing. In addition, both neutrophils and macrophages, which are the main players in innate immune responses, are suggested to have close molecular and functional relationships in the healing processes.
3.2.3 Microglia and Macrophages/Monocytes
Macrophages derived from circulating hematogenous monocytes subsequently enter the lesion, either during the period extending from hours to 3 days after SCI or coincidentally with neutrophils (Fleming et al. 2006; Popovich et al. 1997; Stirling and Yong 2008) (Fig. 3.1A). Their numbers peak at around day 7 (Kigerl et al. 2006). They are CD45high, CD11b+, GR1+/−, and F4/80+ cells (Pineau et al. 2010; Stirling and Yong 2008). They often stay in the lesion for weeks (Okada et al. 2006; Popovich et al. 1997). The time taken for a 50 % reduction from maximum cell numbers is 55 days in rat SCI (compared with 1.2 days for neutrophils and 1.5 days for lymphocytes), suggesting that macrophages/microglia are the main players engaging in chronic inflammation in the lesion (Pruss et al. 2011).
Microglia are the resident tissue macrophages in the CNS. After injury, microglia can be easily and rapidly activated by injury signals in the brain. For example, injury-released ATP induces migration or extension of processes toward an ATP gradient (Davalos et al. 2005; Popovich and Longbrake 2008). Recent genetic fate–mapping studies indicate that microglia are derived from specific precursors in the yolk sac during early embryonic stages and enter the brain thereafter (Ginhoux et al. 2010; Ueno and Yamashita 2014). Under physiological conditions, circulating monocytes are not recruited to the brain to become microglia (Ajami et al. 2007), and they are a different population even in SCI or an experimental autoimmune encephalitis (EAE) model (Ajami et al. 2011; Shechter et al. 2009). Hence, microglia and macrophages/monocytes should be considered as different populations when we study their roles in SCI. Since microglia and macrophages share common molecular profiles, discriminating between them is still however technically difficult. High CD45 expression in hematogenous monocytes and its low expression in microglia could be a suitable marker to distinguish them, but activated amoeboid microglia can exhibit high CD45 expression and need careful interpretation (David and Kroner 2011; Thawer et al. 2013). Fortunately, recent studies have developed novel methods to discriminate between them, and their specific roles are being revealed. Bone marrow chimeras, parabiosis, and transgenic mice (Lys-EGFP mice expressing EGFP in macrophage/monocytes but not in microglia) have been developed for this purpose (Ajami et al. 2011; Mawhinney et al. 2012; Shechter et al. 2009).
Classical experiments have also been conducted by using depletion and transplantation of microglia/macrophages to understand their role in SCI, although these techniques might not clearly distinguish those two populations. Experiments have indicated different detrimental and beneficial effects on recovery from injury. Activated macrophages were seen close to demyelinating areas, suggesting they have toxic properties in SCI (Blight 1985). Indeed, reduction of monocyte/macrophage infiltration and their inactivation using silica, anti-CD11d antibody, and minocycline all improve recovery (Blight 1994; Gris et al. 2004; Stirling et al. 2004). Selective depletion of circulating monocytes using liposomal clodronate can also preserve neural tissue and enhance recovery (Popovich et al. 1999). The secretion of pro-inflammatory cytokines and other biomolecules (MMP-9, iNOS, and superoxide) can be attributed to their toxic role. Indeed, microglia/macrophages express IL-1β and TNF-α within a day (Longbrake et al. 2007; Pineau and Lacroix 2007), and IL-1 receptor antagonist, soluble tumor necrosis factor receptor (TNFR), TNFR1 KO mice, and TNF-α antibody all reduce injury-induced cell death and improve recovery after SCI (Ferguson et al. 2008; Genovese et al. 2008; Nesic et al. 2001). It can be assumed that appropriate activation of macrophages/microglia is critically important in order to control increased toxicity. Indeed, neuron–microglia/macrophage interactions via Cx3cl1–Cx3cr1 and CD200–CD200R have been shown to suppress the abnormal activation of microglia under physiological conditions. Disruption of this signaling aberrantly activates microglia/macrophages and destroys neural tissue (Cardona et al. 2006; Hoek et al. 2000).
In contrast, some other studies have reported contradictory results. Administration of liposomal clodronate alone does not affect the recovery process (Lee et al. 2011). Furthermore, transplantation of macrophages pre-exposed to peripheral nerves promotes recovery after SCI (Rapalino et al. 1998). The same group used elegant transgenic and chimeric methods to show that adoptive transfer of monocytes promotes recovery, whereas depletion of CD11c-DTR+ chimeric monocytes by injection of diphtheria toxin worsens spontaneous recovery (Shechter et al. 2009). In ischemic brain injury, ablation of presumptive resident microglia in CD11b HSVTK mice further damages the lesion (Lalancette-Hebert et al. 2007). These reports are consistent with an immune-regulatory and supportive role for microglia/macrophages in SCI.
These controversial results imply the existence of subtypes of macrophages and microglia. Macrophages have recently been categorized into classically activated pro-inflammatory M1 and alternatively activated anti-inflammatory M2 subtypes (David and Kroner 2011; Sica and Mantovani 2012). This classification might also apply to microglia. Such diversity may explain the contradicting results among experiments. The Th1 cytokine IFN-γ (interferon gamma) can shift the balance toward the M1 state, which produces pro-inflammatory cytokines (IL-12, IL-23, IL-1β, and TNF-α) and ROS and nitrogen species. On the other hand, Th2 cytokines such as IL-4 lead to M2 differentiation, which inhibits the production of pro-inflammatory cytokines and increases interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), as well as other M2 markers like Arginase 1. Recent detailed analyses of M1/M2 phenotypes in SCI have revealed the existence of both M1 and M2 populations. M1 and subsequent M2 populations are observed in SCI (Kigerl et al. 2009; Thawer et al. 2013). Intriguingly, M1 and M2 monocytes/macrophages are recruited along different routes to the lesion site: M1 cells from the adjacent leptomeninges via chemokine (C-C motif) ligand 2 (CCL2) signaling and M2 cells from choroid plexus–cerebrospinal fluid (Shechter et al. 2013). M1 activation is predominant in the chronic phase and may prolong inflammation, which would lead to toxic effects on neurons (Kigerl et al. 2009). ROS, nitric oxide (NO), cytokines (TNF-α, IL-1β), and MMPs secreted by M1 macrophages would potentially be toxic to neural tissue (Block et al. 2007). On the other hand, M2-like IL-10 and arginase-expressing macrophages are recruited in the margin of the lesion, and they are likely to contribute to the healing process (Shechter et al. 2009). Microglia/macrophages can express growth factors such as brain-derived neutrotrophic factor (BDNF), glial-derived neutrotrophic factor (GDNF), insulin-like growth factor 1 (IGF1), and NT-3 in the developing brain and even within the lesion of SCI (Batchelor et al. 1999; Elkabes et al. 1996; Nakajima and Kohsaka 2004; Rolls et al. 2008; Ueno et al. 2013). Those factors are possibly protective in damaged tissue, but their role and characterization in terms of M1/M2 cells have not yet been clearly examined.
The primary role of microglia/macrophages is phagocytosis. After injury, cellular debris, especially myelin debris, is thought to be one of the key factors inhibiting axonal regeneration (Geoffroy and Zheng 2014). Hence, phagocytosis by microglia/macrophage would be important in this context. Indeed, clearance of degenerating axons by macrophage/microglia appears to be important for subsequent axonal regeneration, at least in vitro (Tanaka et al. 2009). In vivo, microglia in the acute phase and subsequent invading macrophages engage in phagocytosis after SCI (Greenhalgh and David 2014). However, it seems that macrophages/microglia are less efficient at clearance within the CNS compared with the peripheral nervous system (Avellino et al. 1995; Lazarov-Spiegler et al. 1996; Lotan and Schwartz 1994), which may explain why axonal regeneration is limited in the CNS. Hence, an increase in phagocytic activity will potentially be beneficial as a therapy.
In addition to their immune-modulatory and phagocytic effects, macrophages/microglia appear to have a direct role in the regenerative processes of neural circuits, especially in axons. Inhibition of microglia/macrophage activation by minocycline treatment can reduce axonal dieback (Kitayama et al. 2011; Stirling et al. 2004). It has been reported that macrophages inhibit axonal elongation (Busch et al. 2009; Horn et al. 2008), and M1 macrophages and microglia inhibit axonal growth in vitro (Kigerl et al. 2009; Kitayama et al. 2011). Microglia can express axon guidance molecules, such as Slit, Netrin (Wehrle et al. 2005), semaphorin 7A (SEMA7A) (Pasterkamp et al. 2003), and repulsive guidance molecule a (RGMa) (Kitayama et al. 2011) in the injured CNS, which could affect axonal remodeling. We have reported that RGMa expressed in activated microglia inhibits axonal growth in vitro, and minocycline treatment can inhibit RGMa expression resulting in reduced axonal dieback (Kitayama et al. 2011). These results strongly suggest that microglia/macrophages can affect the status of axonal regeneration. Interestingly, microglia can also expresses axonal growth factors such as IGF1 (Ueno et al. 2013), thrombospondin (Chamak et al. 1994, 1995), and neurotrophin-3 (NT-3) (Elkabes et al. 1996), suggesting that they may be able to facilitate axon growth under some conditions. However, there is still no direct evidence in vivo that these factors in macrophages/microglia affect axon regeneration. It will be particularly important to know which state of microglia/macrophage activation produces inhibitory or supportive factors for axonal growth, and whether these factors modulate regenerative responses. Recently developed knock-in mice (e.g., the Cx3cr1–Cre and Cx3cr1–CreER lines specifically expressed in microglia; Goldmann et al. 2013), will be ideal tools for understanding the role of each molecule more clearly in vivo.
Although numerous points remain unclear, M2-skewed cells are in general likely to be supportive for tissue repair in SCI. This raises the possibility that treatments that enhance M2 polarization will promote recovery. In this context, strategies that replenish appropriate microglia/macrophages and stimulate axonal regeneration and repair have been evaluated (Bouhy et al. 2006; Lazarov-Spiegler et al. 1996; Rapalino et al. 1998), and preclinical trials in which autologous activated macrophages are injected into injured spinal cord (ProCord) have been conducted as Phase II studies. Although it is a promising idea, some reports indicate that transplantation of M2 macrophages decreases the number of M2 in the lesions of mice, suggesting that the environment there polarizes the cells to M1 and limits the efficiency of therapy (Kigerl et al. 2009). Further evaluation in terms of M1/M2 polarization and molecular mechanisms is needed in order to utilize macrophage/microglia as a therapy. M2-polarizing drugs could be an alternative way, but it may be more effective to combine the methods to modify the lesion milieu.
3.2.4 T Lymphocytes
T cells show a different pattern of infiltration among species after SCI: biphasic invasion in the first and fourth weeks in rats and a gradual increase toward 10 weeks in mice (Sroga et al. 2003) (Fig. 3.1B). T cells are major components in the adaptive immune response and several types of T lymphocytes – CD8+, CD4+ (Th1, Th2, and Th17), and regulatory CD4+ T cells – are involved here (Liblau et al. 2013). CD4+ T cells are composed of: Th1 cells which activate macrophages and CD8+ T cells by IL-2, IFN-γ, and TNF-α; Th2 cells which promote activation and maturation of B cells by IL-4, 5, and 13; Th17 cells which release IL-17, 21, and 22 and activate or recruit neutrophils; and Foxp3+ regulatory T cells which are thought to limit immune responses. CD8-expressing T cells secrete toxic factors, such as perforin, granzymes, and cytokines to directly destroy abnormal cells, such as infected and tumor cells.
The role of T cells in recovery after SCI are again controversial (Popovich and Jones 2003). Many studies indicate that T cells are detrimental to neural tissue and recovery from SCI. In vitro, T cells have a toxic effect on neurons (Giuliani et al. 2003). Reduction of T cells in nude rats (RAG1 −/−, RAG2 −/−) or nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice, in which maturation of both T and B cells is blocked, and on anti-CXCL10 antibody treatment, which inhibits the recruitment of T cells, ameliorate secondary damage and enhance recovery after SCI or TBI (Fee et al. 2003; Gonzalez et al. 2003; Luchetti et al. 2010; Potas et al. 2006; Wu et al. 2012). It has been demonstrated that T cells are activated by CNS antigens and persist in the tissue (Hickey et al. 1991). Some lines of evidence indicate that these auto-reactive T cells are also toxic. For example, myelin basic protein (MBP)-reactive CD4+ T cells exacerbate the lesion and functional deficits after SCI (Jones et al. 2002, 2004; Popovich et al. 1996b).
In contrast, neuroprotective roles have also been reported. For example, auto-reactive T cells are likely to promote recovery in CNS injuries and degenerative diseases such as Alzheimer’s disease, and the “protective autoimmunity” concept has emerged (Schwartz and Raposo 2014). Indeed, adoptive transfer of myelin-reactive T cells and vaccination with auto-antigen (MBP or Nogo-A) has been shown to protect tissue and improve locomotor function after SCI (Hauben et al. 2000, 2001a, b). These approaches are attractive as therapeutic strategies, but careful interpretation is required since conflicting results have also been reported. These controversies may be attributed to the multiple types of T cells. We recently demonstrated that adoptive transfer of Th1-conditioned lymphocytes, but not Th2 or Th17 cells, could enhance the recovery of motor function after SCI (Ishii et al. 2012, 2013). Although the mechanism underlying this beneficial role is still unclear, expression of IL-10 and neurotrophic factors in transferred T cells and the presence of IL-10 expressing activated microglia/macrophages can be correlated with recovery.
The role of regulatory T cells is still under investigation. One report suggests that loss of regulatory T cells exacerbates SCI (Marcondes et al. 2005), but this needs further evaluation. As for Th17 cells, recent reports demonstrate their deleterious effects in EAE and stroke models (Kebir et al. 2007; Shichita et al. 2009). In SCI, transfer of Th17-conditioned cells worsens behavioral recovery in the acute phase (Ishii et al. 2012), but the detailed function and mechanisms are still unknown. Understanding the roles of each T cell subtype in the pathology of SCI and how these populations can be regulated could lead to the establishment of therapeutic strategies.
Reports have proposed further direct roles for T cells in regeneration. In co-culture experiments, activated natural killer (NK) and CD8+ T cells inhibit neurite outgrowth without detectable neuronal apoptosis in a contact-dependent but antigen-independent manner, whereas activated CD4+ T cells promote neurite outgrowth (Pool et al. 2012). Interestingly, activated T cells can produce neurotrophic factors such as BDNF, NT-3, and GDNF (Ishii et al. 2012, 2013; Kerschensteiner et al. 1999), which could be neuroprotective and facilitate regeneration. We reported that Th1 cells, but not Th2 or naive T cells, can enhance axonal elongation in vitro, and that SEMA4A and IFN-γ are involved in this process (Ishii et al. 2010). Semaphorins were originally described as axon guidance molecules in the nervous system, but it has been revealed that they are largely expressed in immune cells and have diverse functions in immune responses (Takamatsu and Kumanogoh 2012). It will be intriguing to examine whether these immune-derived semaphorins affect regenerative responses in SCI.
In conclusion, further analysis is needed on the roles T cells play in axon regeneration, especially in vivo. It should be noted in this case that both indirect and direct effects of T cells could contribute to resilience. For example, transfer of Th1 cells may slightly enhance the regenerative response of axons, but simultaneous activation of macrophages/microglia may directly affect regeneration (Ishii et al. 2012, 2013). In vitro, T cells can enhance the neuroprotective properties of microglia and astrocytes (Garg et al. 2008; Shaked et al. 2005). Thus, the complex interactions among multiple types of immune cells, and neural cells, need careful investigation.
3.2.5 B Lymphocytes
B cells activated by extracellular antigens produce cytokines and antibodies, which target the injured/infected cells that are removed by macrophages. In SCI, B cells appear after a week and chronically persist in the lesion (Ankeny et al. 2006; Schnell et al. 1999a) (Fig. 3.1B). The role of B cells in the repair process remains controversial. In particular, auto-antibodies produced post injury are a focus of debate. Auto-antibodies to MBP, monosialotetrahexosylganglioside (GM1) gangliosides, and myelin-associated glycoprotein (MAG), have actually been detected in human SCI patients (Hayes et al. 2002; Mizrachi et al. 1983; Skoda et al. 2006). Even in mouse SCI, B cells are activated in lymphoid tissues such as those of the spleen, and antibodies are produced against antigens from both inside and outside the CNS (Ankeny et al. 2006, 2009). Although the mechanisms underlying the activation of auto-reactive B (and T) cells are still unclear, cells showing self-recognition are likely to exist as a suppressive mechanism even under physiological conditions (Schwartz and Raposo 2014). Regarding the negative effects, B cell knockout mice show increased amounts of spared tissue and better functional recovery than wild-type (WT) mice (Ankeny et al. 2009). Furthermore, injection of antibodies produced in SCI mice into the spinal cord causes dramatic loss of neuronal cells and aberrant inflammation by macrophages/microglia, accompanied by hind limb paralysis (Ankeny et al. 2009). These results indicate the dark side of B cells and auto-reactive antibodies. As already discussed regarding T cell protective autoimmunity, self-reactive antibodies may play some beneficial roles in recovery, however. For example, B cells produce anti-myelin proteins, which are generally thought to inhibit axonal regeneration. Pre-immunization with spinal cord homogenates promotes axonal regeneration without detrimental immune responses (Huang et al. 1999). Myelin-reactive antibodies are thought to mediate these effects, since antisera containing these antibodies can block inhibition of neurite outgrowth by myelin in vitro.
3.2.6 Glial and Fibrotic Scar
Following inflammatory reaction, other components such as astrocytes and fibroblasts become involved in the creation of scar tissue (Fig. 3.1C). Obviously, activation of astrocytes and the formation of a glial scar are required for proper recovery, since ablation or inactivation of astrocytes using genetically modified mice severely exacerbates inflammation and secondary damage, and affects the recovery process (Faulkner et al. 2004; Okada et al. 2006). They play a role in separating the inflammatory response from neighboring intact neural tissue (Wanner et al. 2013). At the expense of this healing role, they are classically considered a principal barrier to axonal regeneration by expressing chondroitin sulfate proteoglycans (CSPGs) (Silver and Miller 2004). Recent reports, however, suggest that there are several subtypes of astrocytes, some of which can support recovery. For example, TGF-α overexpression increases the numbers of astrocytes that are supportive of axon growth (White et al. 2011), and nuclear factor kappaB (NF-κB) inhibition in astrocytes facilitates the healing process, resulting in reduced lesion volume (Brambilla et al. 2005). Transplantation of astrocytes differentiated from embryonic precursor cells improves recovery and promotes the regeneration of axons (Davies et al. 2006).
Fibroblasts also accumulate in the core of the lesion, where they stabilize the damaged tissue. Although it was believed that these cells are mainly derived from meningeal cells, recent studies using transgenic mice and lineage tracing have demonstrated that the majority of the cells are derived from Glast+ or Col1α1+ pericytes or perivascular cells (Goritz et al. 2011; Soderblom et al. 2013). Importantly, ablation of these populations using Glast-Rasless mice leads to the formation of a large cavity, indicating that fibrotic scar tissue is indispensable for repair (Goritz et al. 2011). On the other hand, the fibroblastic scar is now considered to be the major barrier to axonal regeneration. In the adult CNS, many inhibitory molecules such as myelin-associated proteins and astrocyte-derived CSPGs in the local environment are thought to inhibit axonal regeneration. Moreover, loss of the intrinsic capacity of neurons to regrow axons has been recognized as a second factor impeding regeneration in the adult CNS (Liu et al. 2011). Deletion of Pten, which usually suppresses the mammalian target of rapamycin (mTOR) and AKT pathways, can reactivate this intrinsic signal and induce robust regeneration of axons post SCI (Liu et al. 2010; Zukor et al. 2013), most likely by overcoming most of the inhibitory molecules in the local environment. However, even under these conditions, axons can only grow along bridging astrocytes in the scar, but not over the fibronectin+ fibrotic tissue (Zukor et al. 2013). This strongly suggests that the fibrotic scar is the ultimate barrier to regeneration. Hence, modulation of this structure without weakening the process of repair will be needed to enhance regeneration. From this point of view, an increase in bridging growth-supportive astrocytes to accelerate the process of scar formation (Brambilla et al. 2005; Davies et al. 2006; White et al. 2011), and direct reprogramming of scar cells (Su et al. 2014) are expected to be promising approaches. Transplanting neural cells could be an alternative way to fill the lesion gap, which would simultaneously reconstruct the neural connection (Abematsu et al. 2010; Lu et al. 2012). In addition, these transplanted cells might be able to modulate immune function and lead to better recovery (Pluchino et al. 2005; Uccelli et al. 2007). Immune cells would also affect scar formation, possibly via M2 macrophages/microglia expressing TGF-β. This scar–immune interaction also needs to be examined in more detail.
3.2.7 Targeting Immune to Neural Actions: Future Directions
Since multiple players including immune and glial cells engage in repair processes by communicating with each other, research such as that described here on the role of each cell type and their interactions (immune–immune, immune–glia, immune–nervous) should be undertaken. The glucocorticosteroid methylprednisolone is the only agent widely used in the clinic to suppress the global immune reaction post SCI, but the effect of this treatment is still doubtful and controversial (Jones et al. 2005; Popovich and Longbrake 2008). Thus, it is critically important to understand the complicated neurotoxic and reparative effects of the inflammatory response in order to identify target molecules or cells. In general, a limited inflammatory response would be beneficial for repairing the tissue, but prolonged and excessive inflammatory responses are thought to be deleterious. Hence, therapeutic strategies that accelerate the immune and repair process, and make the lesion smaller, would be the best options for tissue sparing and the reorganization of neural circuits that will lead to efficient recovery.
3.3 CNS Regulation of the Immune System Affects the Recovery Process Post SCI
3.3.1 CNS Regulation of the Immune System
Regarding the brain–immune interaction in CNS diseases and injuries, we also have to consider the nervous system that autonomically regulates immune responses and further influences the recovery process. It has already been highlighted that the immune system is regulated by the nervous system (Fig. 3.2A). The state of the immune system is sensed by the autonomic nervous system via the afferent vagus nerve (Goehler et al. 2000; Tracey 2009), or directly by CNS neurons that are mainly found in circumventricular organs lacking a BBB (Buller 2001) and in the medial preoptic area, via pro-inflammatory cytokines (TNF-α, IL-1β, etc.). Such signals received by the CNS generally cause fever and anti-inflammatory immunosuppressive reactions. In fact, intracerebral or peripheral administration of these cytokines is perceived by neurons and induces physiological changes such as fever, decreased activity, and activation of the stress response by the hypothalamo–pituitary–adrenal (HPA) axis (Tracey 2009). Signals received in the CNS are processed into three output pathways: the HPA axis, sympathetic nervous system (SNS), and parasympathetic nervous system.
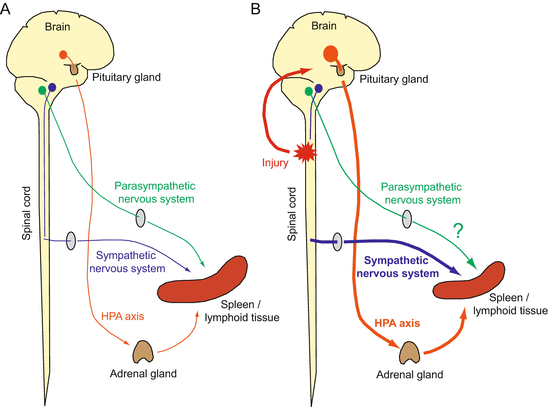
Fig. 3.2
CNS regulation of immune system (brain–immune system interaction): (A) three output pathways – HPA axis (orange), sympathetic nervous system (SNS; blue), and parasympathetic nervous system (green) – are known to be immunosuppressive in regulating immune system homeostasis; (B) following SCI, injury signals (red) are transmitted to the CNS and activate the HPA axis. If the SNS circuit between the brain and spinal cord is destroyed, the local SNS circuit innervating the spleen (or lymphoid tissue) is also activated. Both systems suppress immune reaction. CIDS might be caused by excess response of these inflammatory reflex circuits. The parasympathetic nervous system has not been examined for this condition yet (see the text for details)
In the HPA axis, the signals are first relayed to the paraventricular nucleus (PVN) in the hypothalamus (Haddad et al. 2002). PVN neurons then release corticotropin-releasing factor (CRF), which stimulates the release of adrenocorticotropic hormone (ACTH) in the anterior lobe of the pituitary gland. ACTH further stimulates glucocorticoid secretion in the adrenal gland. Glucocorticoid has a global immunosuppressive effect, by suppression of cytokines (IL-1β, TNF-α, IL-8), prostaglandin, and NO production. It also has apoptotic effects on T lymphocytes and decreases antigen representation.
The SNS is known to innervate lymphoid tissues (thymus, bone marrow, spleen, lymph nodes) and releases norepinephrine (NE) (Felten 2000). NE is able to inhibit pro-inflammatory responses (IFN-γ in Th1 cells; TNF-α and IL-1β in monocytes; NK cell activity), and increase IL-10 expression in monocytes (Meisel et al. 2005). In the parasympathetic nervous system, the immune signals received are reflexively transmitted to peripheral organs through the vagus nerve, and this circuit also has an immunosuppressive role. Indeed, stimulation of the vagus nerve can suppress TNF-α release in peripheral organs, with increased acetylcholine in the spleen. Acetylcholine decreases the release of TNF-α, IL-1β, IL-6, and IL-18 from macrophages (Borovikova et al. 2000). Acetylcholine-synthesizing T cells mediate this vagus nerve–initiated immune suppression (Rosas-Ballina et al. 2011).
3.3.2 Immune Suppression Post CNS Injuries
Infections are a major cause of morbidity and mortality in patients with CNS injuries such as stroke, TBI, and SCI (Meisel et al. 2005). Although these infections occur through invasive treatment, aspiration, and bladder dysfunction, it has been recognized that secondary immunodeficiency caused by CNS injuries (CNS injury-induced immunodepression syndrome, CIDS) underlies the increased susceptibility to infections (Meisel et al. 2005). In SCI, infections such as pneumonia and urinary tract infections are frequently seen in about 28–38 % cases, and about 50 % of deaths are due to infection (Meisel et al. 2005). The immune system is actually suppressed in this condition; the functions of lymphocytes and monocytes are somehow impaired. Immune suppression and vulnerability to infection are frequently observed in stroke, TBI, and SCI patients, depending on the lesion site and volume (Campagnolo et al. 2000; Hagen et al. 2010; Quattrocchi et al. 1992; Riegger et al. 2009). Immune suppression can also be detected in stroke or SCI in rodent models (Offner et al. 2006; Oropallo et al. 2012; Zhang et al. 2013).
Related posts:
 Parkinson’s Disease; Neurodegeneration as Systemic Disease
Parkinson’s Disease; Neurodegeneration as Systemic Disease
 Brain–Peripheral Organ Communication
Intermediate Phenotype Approach for Neuropsychiatric Disorders
Brain–Peripheral Organ Communication
Intermediate Phenotype Approach for Neuropsychiatric Disorders
 Clinical Systems Neuroscience
Clinical Systems Neuroscience
 Home- and Community-based Medical Care for Neurodegenerative Diseases: ALS as an Illustration
Home- and Community-based Medical Care for Neurodegenerative Diseases: ALS as an Illustration
 Significance of Mechanism-Oriented Research Toward Neuronal Protection Therapy Against Neurodegenerative Disorders ~ ZNRF1 E3 Ubiquitin Ligase as a Critical Mediator for Wallerian Degeneration and Neuronal Apoptosis
Significance of Mechanism-Oriented Research Toward Neuronal Protection Therapy Against Neurodegenerative Disorders ~ ZNRF1 E3 Ubiquitin Ligase as a Critical Mediator for Wallerian Degeneration and Neuronal Apoptosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


