, Jean Paul G. Vonsattel2, Helmut Heinsen3, 4 and Horst-Werner Korf5
(1)
Dr. Senckenbergisches Chronomedizinisches Institut, Goethe University Frankfurt, Frankfurt, Germany
(2)
Medical Center Neurological Institute, Columbia University, New York, NY, USA
(3)
Division Psychiatic Clinic Morphological Brain Research Unit, Julius Maximilians University Würzburg, Würzburg, Germany
(4)
University of Sao Paulo Medical School, Sao Paulo, Brazil
(5)
Dr. Senckenbergisches Chronomedizinisches Institut, Goethe University, Frankfurt, Frankfurt, Germany
3.1 Establishment of Cortical Neuronal Loss as a Main Neuropathological Hallmark of Huntington’s Disease (HD) and Its Clinical Relevance
As described in Chap. 2, the neuropathological hallmark of Huntington’s disease (HD) is progressive striatal degeneration, which according to the Vonsattel grading system can be classified into five grades in ascending order of severity of striatal degeneration (Vonsattel 2008; Vonsattel and DiFiglia 1998; Vonsattel et al. 1985). While the affection of the striatum is acknowledged as neuropathological feature of HD since more than a hundred years, the affection of the cerebral cortex during the course of HD and its relevance for the clinical picture were a matter of debate for a long time (see Chaps. 1 and 2). However, a large variety of quantitative and qualitative postmortem studies repeatedly demonstrated cortical neuronal loss in HD patients, which is commonly most pronounced in the isocortical neuronal layers III, V, and VI (Brodmann 1909; Bruyn 1968; Bruyn et al. 1979; Estrada-Sanchez and Rebec 2013; Fennema-Notestine et al. 2004; Forno and Jose 1973; Hedreen et al. 1991; Heinsen et al. 1992, 1994, 1999; Heinsen and Rüb 1997; McCaughey 1961; Richardson 1990; Selemon et al. 2004; Sotrel et al. 1991; Vonsattel 2008; Vonsattel and DiFiglia 1998).
The findings of these postmortem studies eventually allowed to establish the affection of the cerebral cortex as a main neuropathological feature of HD (Figs. 3.1, 3.2, and 3.3), which is currently regarded as the main morphological correlate of psychiatric symptoms, neuropsychological deficits, cognitive decline, and neurophysiological abnormalities of HD patients and some at-risk gene carriers. According to current pathophysiological ideas, the affection of the cerebral cortex not only represents the morphological correlate of these psychiatric, neuropsychological, and neurophysiological manifestations in HD but may at least be jointly responsible for the occurrence of involuntary choreatic movements (Sheperd 2013). Choreatic movements representing the primary involuntary movement abnormalities of HD patients evolve insidiously during the transition from the asymptomatic to the symptomatic phase of HD. They increase and become generalized, abrupt, and uncontrolled only in a second clinical HD stage and then ultimately plateau and wane during the later disease course of HD (Estrada-Sanchez and Rebec 2013; Margolis and Ross 2003; Vonsattel and DiFiglia 1998; Walker 2007a, b). The pathognomonic choreatic movements very frequently are preceded during the prediagnostic phase by a spectrum of psychiatric and/or neuropsychological HD symptoms. Subsequent to their onset, psychiatric and neuropsychological HD symptoms commonly progress concomitantly with the worsening of choreatic movements and finally culminate in severe psycho-organic syndromes or dementia (Estrada-Sanchez and Rebec 2013; Margolis and Ross 2003; Shoulson and Young 2011; Vonsattel and DiFiglia 1998; Walker 2007a, b). Owing to the very early onset of these progressive nonmotor HD symptoms, it is widely believed that the affection of the cerebral cortex may take place even before the striatum undergoes neurodegeneration (Estrada-Sanchez and Rebec 2013; Margolis and Ross 2003). Traditionally, the occurrence of chorea has been widely regarded as an immediate consequence of striatal degeneration (Estrada-Sanchez and Rebec 2013; Mink 1996; Walker 2007a, b), but already Cécile and Oskar Vogt challenged this opinion and assumed that the cortical pathology may play a substantial pathophysiological role in the evolution of chorea (Vogt and Vogt 1920). The traditional understanding of the nature and function of the striatum has been recently unmasked as a circular argument derived from clinical observations (Walker 2007a). Since comprehensive analysis revealed that lesions of the caudate nucleus, putamen, or pallidum result in choreatic movements only in the minority of affected individuals (Bhatia and Marsden 1994), the manifestation of chorea is currently supposed to result from dysfunctional corticostriatal projection neurons and/or their impaired intra-telencephalic input in HD (Estrada-Sanchez and Rebec 2013; Sheperd 2013).
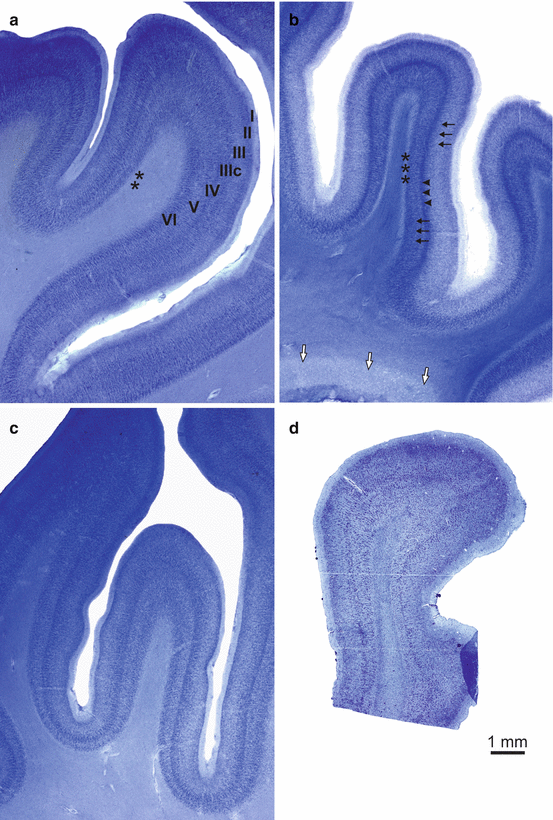
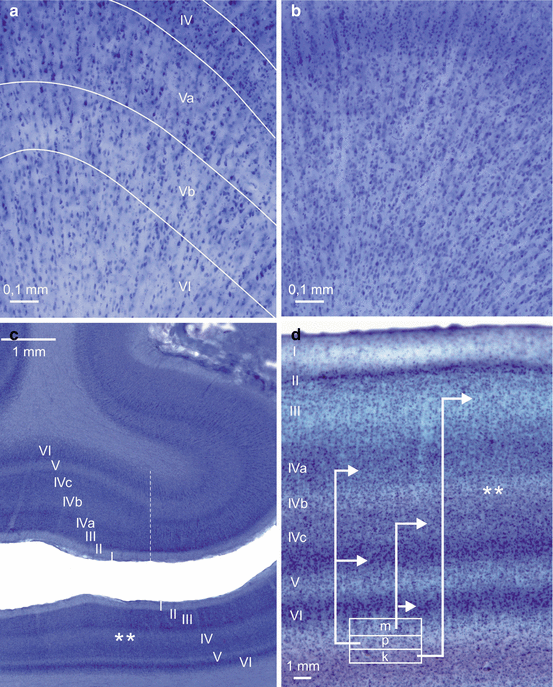
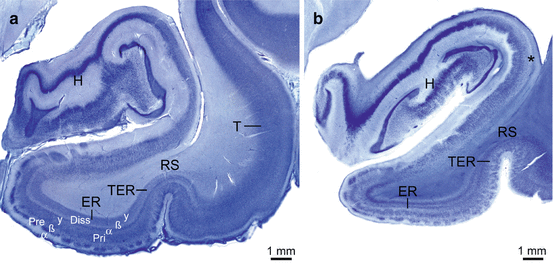
Fig. 3.1
Laminar neuronal loss and white matter changes in the temporal neocortex of Huntington’s disease (HD) patients. (a) Thick stained frontal tissue sections through the occipitotemporal neocortical region (Brodmann’s area 37 or area PH according to von Economo and Koskinas) of a representative control individual, (b) a typical Huntington’s disease (HD) patient, and (c) a typical Alzheimer’s disease (AD) patient (Brodmann 1909; von Economo and Koskinas 1925). (d) 12 μm-thick gallocyanin-stained paraffin section parallel to the thick section shown in (b). (b) Neuronal loss and pallor of pyramidal cell layers III and V (black arrows) and a distinct well-preserved and prominent layer IV (arrowheads) are pathognomonic not only for the temporal association fields but also for all isocortical association fields in the frontal, parietal, and occipital lobes of HD patients (Heinsen et al. 1994). In gallocyanin-stained sections, the cerebral white matter of HD patients commonly shows an intensive staining (i.e., chromophilia). The white matter chromophilia (white arrows) in HD could reflect ongoing attempts of remyelination (Bartzokis et al. 2007) and may be linked to the increased oligodendroglial density in the caudate nucleus of early- and low-grade HD patients (Gómez-Tortosa et al. 2001; Myers et al. 1991). The medullary layer abutting on layer VI and most likely comprising associative u-fibers (asterisks) is intact in HD patients and stains similarly like those of the control individuals. (c) Less conspicuous neuronal loss in layers III and VI and more pronounced neurodegeneration in layer V of the occipitotemporal neocortical region of the AD patient. (d) Whereas unconventionally thick brain tissue sections facilitate diagnosis of cortical neuronal loss in HD, laminar neurodegeneration is difficult to assess in conventional 5–20 μm-thick paraffin sections. Scale bar in D is also valid for a–c. (a–c, gallocyanin staining, 440 μm gelatine sections; d, gallocyanin staining, 12 μm paraffin section)
Fig. 3.2
Laminar nerve cell loss in the temporal neocortex and the primary visual area of the occipital lobe in Huntington’s disease (HD). (a) Unconventional thick frontal tissue sections through the infragranular cortical layers in the crown of the posterior parts of the middle temporal gyrus covered by Brodmann’s area 37 (corresponding to area PH of von Economo and Koskinas) (Brodmann 1909; von Economo and Koskinas 1925) of a typical control case and (b) a representative Huntington’s disease (HD) patient. (a) Note the striking columnar arrangement of the cortical neurons and their separation by unstained radially coursing fiber bundles. (b) Owing to neuropil shrinkage, the surviving layer IV, V, and VI neurons in Brodmann’s area 37 of the HD patient are more closely spaced. Although the total neuronal number is reduced in these cortical nerve cell layers, the apparently higher neuronal density, particularly in layer IV, suggests unchanged neuronal numbers thereby masking true neuronal loss. (c) Thick frontal tissue section through the caudal pole of the occipital lobe of a typical control individual at the border between Brodmann’s primary visual area 17 and the adjacent parastriate Brodmann’s area 18 (dashed vertical line). The “calcarine cortex type” of Brodmann’s primary visual area 17 is characterized by: (1) a striking reduction of the outer and inner pyramidal layers III and V, (2) conspicuous granular layers II and IV with a particularly high neuronal density, (3) a strongly basophilic multiform layer VI, and (4) the subdivision of the inner granular layer into layers IVa–c. Note the prominent pale, cell-poor, and myelin-rich Gennari stripe in the inner granular layer IVb (asterisks) (Brodmann 1909; Garey 1990; Rüb et al. 2014b; Zilles 1990). (d) High-power photograph of a thick tissue section through the degenerated primary visual area 17 of a typical HD patient: (1) severe nerve cell loss and pallor of the outer pyramidal layer III, (2) markedly reduced nerve cell density and pallor of the inner granular layers IVa and IVc, (3) narrowed multiform layer VI reflecting nerve cell loss. Asterisks indicate Gennari stripe (Rüb et al. 2014b). Since Brodmann’s primary visual area 17 has no direct anatomical interconnectivities with the striatum, its layer-specific degeneration in HD most likely is not the result of a retrogradely spreading neuronal loss following the initial degeneration in the striatum but may be a primary pathological event. White box in (d) symbolizes the six-layered thalamic lateral geniculate body (m magnocellular layer, p parvocellular layer, k koniocellular layer) from where the granular layers II and IV and the pyramidal layer III of Brodmann’s primary visual area 17 receive visual projections via the optic radiation. Arrows mark the laminar targets of these subcortical projections in Brodmann’s primary visual area 17 (Garey 1990; Rüb et al. 2014b; Zilles 1990). (a–d: gallocyanin staining, 440 μm gelatine sections). (d: Modified according to Rüb et al. (2014b), (Figure 1, page 169); with kind permission from John Wiley and Sons). Abbreviations: m magnocellular layer of the thalamic lateral geniculate body, p parvocellular layer of the thalamic lateral geniculate body, k koniocellular layer of the thalamic lateral geniculate body
Fig. 3.3
Laminar neuronal loss in the allocortical transentorhinal and entorhinal regions in Huntington’s disease (HD). (a) Thick gallocyanin-stained frontal section through the parahippocampal gyrus and hippocampus of the left temporal lobe of a typical control individual and (b) a representative Huntington’s disease patient. (b) Moderate neuronal loss in the subiculum of the HD patient (asterisk) and nearly complete degeneration of the pre-β and pri-γ layers in the entorhinal (ER) (Brodmann’s area 28) and transentorhinal (TER) (Brodmann’s area 35) regions (Braak and Braak 1992a, b; Brodmann 1909; Heinsen et al. 1992). (a, b: gallocyanin staining, 440 μm gelatine sections). Abbreviations: Diss lamina dissecans, ER entorhinal cortex, H hippocampus, Pre principal external layer, Pri principal internal layer, RS rhinal sulcus, T temporal neocortex, TER transentorhinal cortex
3.2 Confirmation and Extension of Very Early Descriptions of Cortical Neurodegeneration in Huntington’s Disease (HD) by Modern Quantitative Approaches
Initial conjectures, qualitative assessments, and the first quantitative attempts around the turn of the century already suggested degeneration and nerve cell loss in the cerebral cortex of HD patients (see Chap. 1). However, it was only in the 1980s when the first quantitative postmortem investigations of the brains of HD patients were performed by means of more sophisticated methods. According to our and others’ experience, the true nerve cell loss in a given brain region can be unequivocally detected upon qualitative light-microscopical investigation only, if its extent exceeds a threshold of approximately 60 %. To investigate early neurodegenerative events on a solid base and to obtain insight into neurodegenerative events that are less severe than those in the striatum, quantitative morphometric methods were applied to assess volumes, cross-sectional areas, and widths, as well as cell densities in the cerebral cortex and associated white matter in the brains of HD patients.
The first pioneering and groundbreaking volumetric studies of serial tissue sections through the brains of HD patients yielded atrophy and volume loss of distinct cortical areas and regions. Cortical volume loss was 30 % in the peristriate and parastriate visual areas in the occipital lobe (i.e., Brodmann’s areas 18 and 19), 17 % in the primary visual striate area covering the calcarine sulcus in the occipital lobe (i.e., Brodmann’s area 17), 19 % in the primary somatosensory cortex (i.e., Brodmann’s areas 1, 2, and 3), 12–21 % in the frontal cortex, 15 % in the anterior cingulate cortex (i.e., Brodmann’s area 24) and 25 % in the posterior cingulate cortex (i.e., Brodmann’s area 23), 12 % in the primary auditory cortex (i.e., Brodmann’s areas 41 and 42), 12 % in the insula, 10 % in the temporal neocortex, and 10 % in the parahippocampal gyrus with the transentorhinal and entorhinal regions (Brodmann 1909; Lange 1981; Lange and Aulich 1986). In addition to detailed findings in the cerebral cortex, these accurate morphometric studies also showed degenerative signs in subcortical brain regions of HD patients, which stimulated further quantitative HD studies that directly or indirectly confirmed the initial observations in cortical and subcortical regions (Fig. 1.4). Taken together, the findings of these accurate morphometric studies contrasted with the traditional reductionistic viewpoint of HD as a degenerative disease predominantly affecting the striatum and pointed to a widespread but area-specific vulnerability of the cerebral cortex in HD. Representing milestones of neuropathological HD research, these groundbreaking morphometric studies have definitely changed the traditional unilateral pathoanatomical and pathophysiological concepts of HD, which solely took the affection of the striatum into consideration. They contributed substantially to the paradigm shift in neuropathological HD research, which ultimately paved the way for the breakthrough of the concept of HD as a multisystem neurodegenerative disease (Heinsen et al. 1994; Lange and Aulich 1986; Rüb et al. 2013a; Vonsattel 2008).
Estimations of brain weights disclosed a reduction of the total brain weight by 30 % in the HD individuals studied, and evaluation of brain cross-sectional areas found a mean reduction of the cross-sectional area of the cerebral cortex by 21–25 % and of the underlying cerebral white matter by 29–34 %, both of which correlated negatively with higher grades of HD severity (De la Monte et al. 1988). Ten years later the results on the decline of brain weight and reduction of the cross-sectional areas of the cerebral cortex and white matter of HD brains were confirmed and extended by the findings of a reduced length and thickness of the cerebral cortex or some of its areas in HD. A comparison of these morphometric postmortem results suggested that loss of cerebral white matter in the brains of HD patients is more severe than the reduction of cerebral gray matter (Mann et al. 1993). In 1991, Hedreen et al. reported a statistically significant decrease of the total cortical thickness and widths of cortical layers III, V, and VI of the dorsal bank of the superior frontal sulcus in HD patients which belongs to Brodmann’s cortical area 10 (Brodmann 1909; Hedreen et al. 1991).
Further analysis of the brain volumes pointed to a widespread and global disease process in HD patients and demonstrated severe brain atrophy resulting from volume reductions in both cortical and subcortical gray matters (e.g., striatum). Although the cortex of the medial temporal lobe seemed to be more or less spared, cortical atrophy was relatively uniformly distributed over the four cerebral lobes (Fig. 2.10). Striatal atrophy correlated with the severity of cortical atrophy and loss of frontal white matter with both cortical and striatal atrophies (Halliday et al. 1998).
Reconsiderations of the theoretical background of the quantitative morphometric studies that were widely appreciated and applied in basic neurological and psychiatric research at that time led to the insight that estimations of volumes, widths, or cross-sectional areas of distinct brain regions may suffer from bias due to complex agonal, postmortem, and technical factors (e.g., brain tissue shrinkage). These factors can influence the morphometric parameters in an unpredictable manner; they may reflect atrophy of nerve cells, their processes, and/or other components of the brain neuropil in a false-positive or false-negative way. Since they cannot be effectively compensated by strict protocols of brain fixation, brain embedding, and staining procedures (Coggeshall and Lekan 1996; Heinsen and Heinsen 1991; Heinsen et al. 1992, 1994, 1996, 1999; Oorschot 1994; Schmitz and Hof 2005; West and Gundersen 1990), analyses without correction of the bias do not necessarily provide data that reliably substantiate true death of nerve or glial cells. As in other fields of neuroscience, estimations of volumes, widths, or cross-sectional areas of distinct brain regions were therefore complemented and replaced by morphometric estimations of neuronal densities in neuropathological HD research. These quantitative estimations of neuronal densities, however, have been regarded only for a limited time span as morphometric parameters that can furnish additional and better quantitative arguments in the assessment and diagnosis of brain atrophy and/or neurodegeneration (Coggeshall and Lekan 1996; Heinsen and Heinsen 1991; Heinsen et al. 1992, 1994, 1996, 1999; Oorschot 1994; Schmitz and Hof 2005; West and Gundersen 1990).
One of the earliest morphometric postmortem studies which was based on the estimation of nerve cell densities described a significant reduction of the density of pyramidal projection neurons immunoreactive for nonphosphorylated neurofilament proteins (SMI-32) and reported a normal density of cortical nerve cells immunoreactive for the neuropeptide Y (NPY) in Brodmann’s areas 8, 9, 11, and 22 of HD patients (Brodmann 1909; Cudkowicz and Kowall 1990). Subsequently, the reduction of the density of SMI-32 immunoreactive nerve cells was also proven in the angular gyrus of HD patients located at the junction of the temporal, parietal, and occipital lobes (i.e., Brodmann’s area 39) (Brodmann 1909; MacDonald et al. 1997).
Based on stereological countings of various cortical cell types (i.e., large pyramidal neurons, small neurons, astrocytes, and oligodendroglial and microglial cells), recording of the thickness of each neuronal layer of the cerebral cortex, and estimations of nerve cell densities, a detailed postmortem study performed on Brodmann’s prefrontal areas 8 and 9 of HD patients extended the findings of Cudkowicz and Kowall and demonstrated a selective and progressive loss of a subset of the large pyramidal neurons in cortical layers III, V, and VI, which was accompanied by a decrease in the thickness of these cortical neuronal layers. As in previous and subsequent HD studies, degeneration of cortical pyramidal neurons was interpreted either as a primary event in the pathological process of HD or as the result of a retrograde process induced by a primary degeneration in the striatum (Sotrel et al. 1991).
An additional quantitative postmortem HD study employed three-dimensional cell counting methods for the assessment of laminar cell densities, cell numbers in select cortical areas, and laminar widths. This study discovered an overall reduction of the cortical thickness of Brodmann’s areas 9 (26 %) and 46 (23 %); a reduction of the density of projection neurons in layers III (16 %), V (31 %), and VI (37 %); and a reduced width of these layers (III–25 %, V–34 %, and VI–46 %) in Brodmann’s area 9. In contrast, reduction of the width of cortical layers II (18 %) and VI (35 %) was not accompanied by neuronal loss. In addition, the density of glial cells was significantly increased in layer VI (68 %) of Brodmann’s area 9 and in layer V (75 %) of Brodmann’s area 46. In view of the disparity in prefrontal cortical neurodegeneration, the authors suggested that (1) the prefrontal neurodegeneration underscores the dissociation of cortical neuronal loss from striatal retrograde degeneration, (2) the affection of the cerebral cortex in HD may not result from retrograde degeneration induced by the striatal pathology, (3) retrograde corticostriatal degeneration is not the driving force behind the cortical pathology in HD, and (4) the pathological changes in the cerebral cortex are produced by a cascade of downstream neurodegeneration in the cortico-striato-thalamocortical loops and eventually triggered by neurodegeneration of thalamic nuclei (Brodmann 1909; Selemon et al. 2004).
These quantitative postmortem HD studies confirmed and extended the early findings about the state of the cerebral cortex in HD patients from the 1990s, provided additional evidence for an overall neuronal loss in the cerebral cortex and the differential vulnerability of its distinct areas, and supported the arising opinion that degeneration of the cerebral neo- and allocortex may be among the neuropathological hallmarks of HD and of substantial relevance for the clinical picture of HD.
3.3 Application of Advanced, Unbiased Stereological methods for the Assessment of Nerve Cell Loss
The field of quantitative brain research methods, however, progressed further. Approximately 20 years ago, additional efforts culminated in the design and implementation of sophisticated stereological methods that facilitate unbiased estimations of total cell numbers, which are not confounded by agonal, postmortem, or technical factors and therefore reflect their real extent in the brain region studied (Coggeshall and Lekan 1996; Heinsen and Heinsen 1991; Heinsen et al. 1992, 1994, 1996, 1999; Oorschot 1994; Schmitz and Hof 2005; West and Gundersen 1990).
Related posts:
 Pathological Nerve Cell Alterations in Huntington’s Disease (HD) and Their Possible Role for the Demise of Nerve Cells
Conclusions and Outlook
Pathological Nerve Cell Alterations in Huntington’s Disease (HD) and Their Possible Role for the Demise of Nerve Cells
Conclusions and Outlook
 Widespread Brainstem Neurodegeneration in Huntington’s Disease (HD)
Widespread Brainstem Neurodegeneration in Huntington’s Disease (HD)
 Intraneuronal Transport and Defense Mechanisms with Possible Pathogenetic Relevance in Huntington’s Disease (HD)
Intraneuronal Transport and Defense Mechanisms with Possible Pathogenetic Relevance in Huntington’s Disease (HD)
 Degeneration of Select Motor and Limbic Nuclei of the Thalamus in Huntington’s Disease (HD)
Degeneration of Select Motor and Limbic Nuclei of the Thalamus in Huntington’s Disease (HD)
 Consistent and Widespread Degeneration of the Cerebellum in Huntington’s Disease (HD)
Consistent and Widespread Degeneration of the Cerebellum in Huntington’s Disease (HD)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


