CHAPTER 86 THE CONGENITAL MYOPATHIES
The congenital myopathies are a clinically and genetically heterogeneous group of congenital muscle disorders with characteristic structural abnormalities evident on muscle biopsy, visible after preparation with specific histochemical stains and/or on electron microscopy. Central core disease (CCD),1,2 nemaline myopathy,3 myotubular (centronuclear) myopathy,4 and minicore myopathy (or multiminicore disease [MmD])5 are the major disease entities. Other conditions with more unusual structural abnormalities are very rare, and it is not clear whether all are genetic entities.6
Autosomal dominant, autosomal recessive, and X-linked inheritance are all recognized in this group of disorders, and some conditions such as nemaline myopathy, CCD, and myotubular myopathy may have more than one mode of inheritance. Genetic advances have implicated several genes encoding sarcomeric and sarcotubular proteins (Table 86-1). The boundaries between these conditions, originally defined according to histopathological and clinical criteria, are often indistinct and do not necessarily reflect underlying molecular mechanisms: Mutations in the same gene can indeed give rise to diverse clinical and histopathological phenotypes, and, conversely, a similar histopathological and clinical phenotype may arise from mutations in a variety of genes. Although clinical management is currently the main form of treatment of the congenital myopathies, further advances in the understanding of the precise molecular mechanisms underlying each disorder may result in more rational therapeutic options in the future.
EPIDEMIOLOGY
Epidemiological data on the congenital myopathies are few, and larger geographical surveys are limited. The overall incidence of the congenital myopathies is estimated at 6 per 100,000 live births, representing approximately 10% of all neuromuscular disorders.7 Studies in northern Ireland8 and western Sweden9 suggest that the prevalence of the congenital myopathies in a pediatric population is between 3.5 and 5.0 per 100,000. The relative frequency of individual conditions is unknown, but CCD and conditions associated with mutations in the skeletal muscle ryanodine receptor gene (RYR1) appear to be more common in the patient population (see later discussion) than are nemaline myopathy and the much rarer centronuclear myopathies. Also, the prevalence of specific congenital myopathies may have been previously underestimated, inasmuch as not all muscle biopsy specimens from individuals carrying disease-causing mutations exhibit the characteristic structural abnormalities.10
CLINICAL FEATURES
Most of the clinical features are nonspecific, despite some variations in overall severity, distribution of weakness, and associated features. The diagnosis of a specific congenital myopathy can therefore be made only tentatively on clinical grounds alone, and muscle biopsy is required for a precise diagnosis in individual cases.
Presentation of the patient is either as a “floppy infant” with generalized hypotonia at birth followed by developmental delay or with weakness of variable severity and distribution later in childhood. Presentation in adulthood has also been described, but other disease processes mimicking the appearance of a congenital myopathy should be considered.11 Presentation at birth is often associated with “myopathic” facial features, feeding difficulties, and respiratory difficulties; thinning of the ribs and chest deformities may indicate antenatal onset. Arthrogryposis may be present in the most severe cases of nemaline myopathy12 and CCD13 but is not a common feature. Most cases manifest later in infancy or childhood with motor developmental delay or predominantly proximal weakness that mimics a limb girdle muscular dystrophy or a mild form of spinal muscular atrophy. In other patients, there is marked weakness of the axial muscles and/or the face; a minority have prominent distal involvement. Extraocular involvement is common in centronuclear myopathy and specific subgroups of minicore myopathy, but not in CCD and nemaline myopathy.
Spinal deformities include exaggerated lordosis, scoliosis, and spinal rigidity.14 Scoliosis may be present at birth but typically manifests in a progressive manner around the time of the pubertal growth spurt. Ligamentous laxity is common. Joint contractures, mainly of the Achilles tendon, tend to progress over time. Tendon reflexes are weak or absent. Congenital dislocation of the hips is a common feature in CCD15 but can occur in several other neuromuscular disorders.
Respiratory involvement secondary to diaphragmatic and/or intercostal muscle weakness is the main prognostic factor.16,17 Respiratory impairment is common in centronuclear myopathy, nemaline myopathy, and subgroups of minicore myopathy; it may occur only rarely in CCD,18 with the exception of the severe congenital variant, in which it is common.13 Susceptibility to respiratory infections is frequent, particularly early in life, but may improve with time.13,19 Cardiac involvement other than cor pulmonale secondary to respiratory impairment is not usually a feature, although structural abnormalities such as mitral valve prolapse have been occasionally documented. However, a few patients have had congenital myopathy with rods and/or cores and cardiac involvement in association with mutations in the skeletal muscle αactin gene (ACTA1).20
There are no associated structural central nervous system or peripheral nerve abnormalities, and intelligence is usually normal. In the few cases in which an autopsy was performed, no central or peripheral nerve abnormalities could be identified.21–23
Progression of muscle weakness in the absence of substantial respiratory impairment does not usually occur, but deterioration is occasionally associated with growth spurts or marked weight gain. The most severely affected neonates may die from respiratory failure, but long-term survival has been reported even in infants with severe hypotonia and marked respiratory impairment.13,19,24,25
INVESTIGATIONS
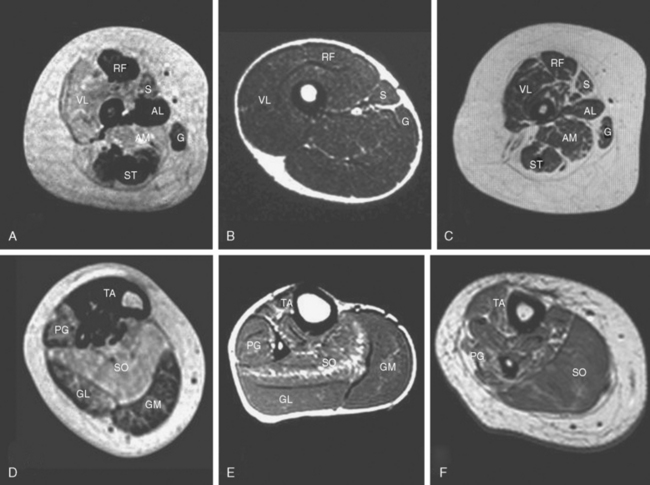
Other investigations may support the suspicion of an underlying myopathic process, but, with the possible exception of muscle imaging, they rarely aid in the diagnosis. Diseased muscle exhibits characteristic changes in echogenicity and signal intensity on ultrasonography, computed tomography, and magnetic resonance imaging (MRI), which reflect an increase in adipose and connective tissue.7,25–27 Muscle MRI in particular may reveal a characteristic pattern of selective involvement in conditions such as CCD and nemaline myopathy, thereby guiding the molecular diagnosis in clinically and histopathologically equivocal cases.28,29
The serum creatine kinase level is usually normal or only mildly elevated. The electromyogram may appear normal in the young patients or in mild cases but usually reveals nonspecific myopathic features, comprising small-amplitude polyphasic potentials.30,31 Additional “neurogenic” changes may be present in distal muscles, particularly of patients with nemaline myopathy.31–33 Spontaneous activity resembling findings in spinal muscular atrophy have been reported in the most severely affected neonates.34
MANAGEMENT
Respiratory impairment is the most important complication and should be anticipated by regular monitoring of respiratory function: regular measurement of forced vital capacity, in sitting and lying positions in order to evaluate diaphragmatic function, and annual overnight oxygen saturation monitoring if the forced vital capacity falls under 60% or, if clinically indicated by symptoms of nocturnal hypoventilation such as early morning headaches, loss of appetite, and daytime tiredness.35 Respiratory impairment may be disproportionately severe, and respiratory failure may occur even in ambulant patients.16,17,19,35–37 Nighttime noninvasive ventilation offers an effective means of improving quality of life and overall prognosis in otherwise only moderately disabled patients. Respiratory infections should be treated aggressively. A cardiac ultrasound study should be performed to document baseline cardiac function and occasionally associated structural cardiac abnormalities. Regular cardiac ultrasonography should be performed in patients with respiratory impairment, although appropriate respiratory management should prevent cor pulmonale.16 Primary cardiomyopathies are rare in the congenital myopathies but have been reported in individual cases with mutations in the ACTA1 gene20 and in a few patients with minicores evident in muscle biopsy specimens (see later discussion).
Spinal posture should be monitored closely and surgical procedures for scoliosis planned carefully to ensure that respiratory function is still sufficient at the time of operative intervention.38 Regular physiotherapy is aimed at preservation of muscle power and prevention of contractures. When orthopedic intervention is planned, the potential benefit has to be weighed against adverse effects of prolonged immobilization.
Patients with CCD and other congenital myopathies associated with mutations in the RYR1 gene are at increased risk of malignant hyperthermia susceptibility,39 and potentially triggering anesthetic agents should be rigorously avoided in these cases. Although a higher anesthetic risk is not clearly documented in other congenital myopathies, similar precautions should be taken in the anesthetic preparation of other patients,35 particularly in those in whom the genetic background is unclear.
Pharmacological interventions have not been widely used in the treatment of the congenital myopathies. A small pilot study has demonstrated some beneficial effect of salbutamol in the treatment of patients with CCD and minicore myopathy,40 but this has yet to be confirmed in a larger controlled trial.
CENTRAL CORE DISEASE
Histopathology
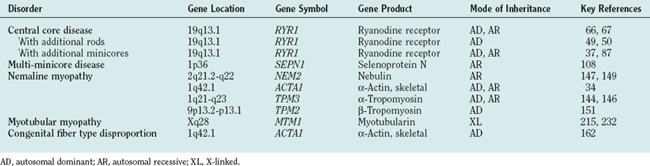
The term central core disease (Mendelian Inheritance in Man [MIM] number 117000)41 was introduced in 195842 after the original description of a family with five affected individuals in three generations who showed amorphous central areas in muscle fibers with the modified Gomori trichrome stain1; absence of oxidative enzyme activity within the core area was subsequently identified as the characteristic histopathological feature (Fig. 86-1A).27 The cores have a predilection for type 1 fibers and extend along a significant length of the longitudinal muscle fiber axis43; localization is characteristically single and central, but multiple, peripheral, and eccentric cores may occur within the same sample.10 In some cases, large typical cores may not be apparent, but only unevenness of oxidative enzyme stain or small core areas are visible, producing a confusing pathological overlap with minicore disease. Pronounced type 1 predominance or uniformity is common and may be the only feature at presentation, particular in very young patients.10,43 The degree of associated increases in fat and connective tissue depends both on the age of the patient and on the sampling site, because the degree of selective muscle involvement may be considerable29; centrally placed nuclei may also be present but are not a consistent feature. Necrosis and regeneration are not usually seen in CCD.
Ultrastructurally, typical cores show a reduction or absence of mitochondria, reflected in the absence of oxidative enzyme staining, accompanied by varying degrees of sarcomeric disruption and accumulation of Z line material.43 In most cases, cores are “structured” with hypercontracted myofibrils that retain adenosine triphosphatase activity but are depleted in mitochondria and sarcoplasmic reticulum; “unstructured” cores, on the contrary, show an absence of adenosine triphosphatase activity, marked disruption of myofibrils, and accumulation of Z line material.
Immunohistochemical studies have demonstrated accumulation of the intermediate filament protein desmin within cores and at their perimeter.10,44,45 Accumulation of other proteins also occurs, including the actin cross-linking protein γ-filamin, αβ-crystallin, small heat shock protein 27, and myotilin46; however, these are nonspecific findings and may be observed in core formation of different etiology.
Core formation is a nonspecific secondary phenomenon and is not correlated with the degree of the muscle weakness. Core formation can be observed after tenotomy,47 in the muscle of patients with long-standing neurogenic atrophy (“target fibers”), and in association with several other gene defects. Central corelike structures may be found in association with dilated cardiomyopathy in patients with mutations in ACTA120 or in the β-myosin heavy-chain gene (MYH7)48; however, in the latter group, there is usually no or little associated weakness. Central core and nemaline rods are occasionally observed in the same biopsy specimen in patients with mutations in the RYR1 gene.49,50
Clinical Features
“Classical” CCD is usually inherited as a dominant trait with a fairly consistent clinical phenotype; sporadic cases with similar clinical features are increasingly recognized. Presentation is in infancy with hypotonia or in early childhood with motor developmental delay or proximal weakness17; however, more severe presentations with features of the fetal akinesia sequence have been reported.13,51 Most of these infants required ventilation from birth, and their course was severe and eventually fatal; however, one infant was eventually weaned off the ventilator and became independently mobile.13
Weakness in most familial cases is pronounced in the hip girdle and in the axial muscle groups17; in rare cases, there is associated muscle wasting.52 Facial involvement is usually mild, and lack of complete eye closure may be the only finding. Bulbar and extraocular involvement is usually absent. Some patients may have prominent exercise-induced myalgia.53
Orthopedic complications are common and comprise congenital dislocation of the hips, foot deformities, and scoliosis.15,18,54 Contractures other than Achilles tendon tightness are rare, and many affected individuals have marked ligamentous laxity, occasionally in association with patellar instability.54
Cardiac abnormalities other than occasional mitral valve prolapse have rarely been reported.55 Respiratory involvement is typically milder than in other congenital myopathies but may be severe in some sporadic and recessive cases.51
Malignant hyperthermia susceptibility (MHS) is a frequent complication39,55,56 and should be anticipated in the anesthetic preparation of patients with CCD. Some patients with MHS may show consistent dysmorphic features (King-Denborough syndrome), including ptosis, down-slanting palpebral fissures, neck webbing, scoliosis, pectus deformity, short stature, and cryptorchism57,58; additional findings in other families may include vertebral fusion, eventration of the diaphragm, and spinal cord tethering.58,59
Except for patients with the most severe neonatal cases and some patients with congenital dislocation of the hips,37,60 most patients achieve the ability to walk independently. The course of CCD is static or only slowly progressive, even over prolonged periods of follow-up.61
The serum creatine kinase level is usually normal or only mildly elevated.62,63 Muscle ultrasonography often shows a striking increase in echogenicity and differential muscle involvement, even in paucisymptomatic individuals.64 Muscle MRI reveals a characteristic pattern of selective muscle involvement in patients with typical CCD29 (Fig. 86-2A and D) and may complement clinical assessment.
Genetics of Central Core Disease and Malignant Hyperthermia Susceptibility
MHS was recognized in 1960 as a familial, autosomal dominant trait by M. A. Denborough and R. R. H. Lovell in Australia and has been linked to several loci. Both CCD and MHS show considerable clinical overlap and have been associated with mutations in the RYR1 gene at chromosome 19q13.1.65–67 CCD is usually inherited as a dominant trait; however, sporadic cases either caused by de novo dominant mutations or associated with recessive inheritance are increasingly recognized.
Although it is now apparent that mutations in the RYR1 gene (MHS1) account for the majority of familial MHS cases,68 locus heterogeneity has been suggested by variable degrees of linkage evidence for several loci (MHS2 to MHS6) and cosegregation of MHS susceptibility with mutations in candidate genes.69–74 There is also the possibility that the MHS phenotype may reflect the compound influence of several genes rather than a major gene defect.75 Genetic homogeneity in CCD has been suggested by RYR1 linkage in most families without confirmed mutation. Although dominant mutations in the ACTA1 gene20 and the MYH7 genes48 may mimic the histopathological appearance, clinical features such as an associated cardiomyopathy are divergent from those in classic CCD.
More than 50 RYR1 mutations have been reported to date in association with CCD, MmD (described later), and MHS with or without cores evident on muscle biopsy specimens.76–80 Most RYR1 mutations are missense mutations; a few small deletions78,79,81 and a cryptic splicing site mutation51 have also been documented. The first reported RYR1 mutations gave rise predominantly to the MHS phenotype and affected mainly two regions of the protein: the cytoplasmic N-terminal domain (MHS/CCD region 1, amino acids 35 to 614) and the cytoplasmic central domain (MHS/CCD region 2, amino acids 2163 to 2458). Although mutations associated with a congenital myopathy phenotype have been identified in these regions,66,67,82,83 there has been increasing evidence that mutations affecting the C-terminal portion of the receptor molecule (MHS/CCD region 3, amino acids 4550 to 4940) are common in patients with CCD.49,78–80,84 Although the mutations are occasionally reported in the C-terminal portion of the protein,85 a series of 124 unrelated North American patients with MHS confirmed the N-terminal and central portion of the protein as the predominant sites of MHS-related RYR1 mutations.86
Although the majority of RYR1 mutations associated with MHS or CCD described to date are dominant missense mutations, recessive inheritance of RYR1 mutations has been reported in mild cases with histopathological features of MmD and CCD37,87 and in a severe form of CCD manifesting with a fetal akinesia syndrome.13 These findings suggest that clinically silent MHS mutations may give rise to a severe phenotype in the compound heterozygous or homozygous state, caused by a combined deleterious effect on the tetrameric RyR1 protein.
The effects of specific RYR1 mutations on excitability and calcium homeostasis have been studied in various homologous and heterologous expression systems. Two models for mutation-induced receptor malfunction have been proposed: depletion of sarcoplasmic reticulum calcium stores with resulting increases in cytosolic calcium levels (the “leaky channel” hypothesis)88 and a specific disturbance of excitation-contraction coupling (the “E-C uncoupling” hypothesis).89 The “ectopic” RYR1 expression in B lymphocytes has been recognized; this offers an easily accessible model for studying the effects of RYR1 mutations in vitro. B lymphocytes harboring CCD-related RYR1 mutations exhibit unprompted calcium release with resulting depletion of sarcoplasmic reticulum stores78,90; increased release of inflammatory cytokines in the same study91 may also indicate a role of RYR1 in immunomodulation.
MULTIMINICORE DISEASE
Multicore disease (MIM numbers 255320 and 157550)41 was originally described in two unrelated children with hypotonia and muscle weakness dating from infancy and a characteristic histopathological appearance of multiple, small, well-circumscribed foci of myofibrillar degeneration with reduction of oxidative enzyme staining.5 Since the original description, at least 100 other cases with similar histopathological features and a wide range of clinical phenotypes have been reported, under different names such as minicore myopathy, myopathy with multiple minicore, pleocore disease, or myopathy with focal loss of cross-striations. The designation multiminicore disease92 used in this chapter reflects the larger number and smaller size of characteristic lesions compared with classic CCD.
Histopathology
Minicores are areas of focal myofibrillar disruption with a variable degree of Z line disorganization and may occur in both type 1 and type 2 fibers (see Fig. 86-1B). They extend only a short distance along the longitudinal axis of the muscle fiber, affecting a variable number of sarcomeres.5,43 In rare cases, the entire fiber diameter may be involved (focal loss of cross-striations).93,94 Pathological distinction between CCD and MmD is occasionally difficult, because in some biopsy specimens from patients with RYR1 mutations, only unevenness of oxidative enzyme stain or minicores are seen, and minicores may evolve into central cores with time.10,87
Commonly associated features are predominance or uniformity of type 1 fibers, often hypotrophic, with or without type 2 hypertrophy.95 This is particularly true for cases secondary to RYR1 but not selenoprotein N, 1 (SEPN1) mutations (see later discussion). Although central nuclei may be a feature in cases caused by RYR1 mutations, they are not usually associated with SEPN1 mutations. The presence of whorled fibers and increases in fat and connective tissue in some affected families43 are suggestive of a potential overlap with the milder congenital muscular dystrophies.
On electron microscopy, minicores are characterized by focal areas of myofibrillar disruption with paucity of mitochondria.43 The sizes of minicores and the degree of myofibrillar disruption are variable, ranging from mild Z line streaming to areas with complete loss of sarcomeric organization. These may be observed in the same biopsy speciment.5 In some areas, ultrastructural abnormalities may consist only of subtle misalignment of myofibrils accompanied by an absence of mitochondria.
In immunohistochemical studies, authors have reported abnormalities on staining with desmin45 and γ-filamin antibodies,46 similar to those observed in CCD.
Minicores are nonspecific; they may be found in various disorders such as muscular dystrophies; denervation; and inflammatory, endocrine, and metabolic myopathies. Also, minicore-like structures may coexist with central cores, central nuclei, and nemaline rods in the same biopsy specimen10,20,96–102 and in association with various gene defects.
Clinical Features
MmD usually manifests in infancy or childhood with hypotonia or proximal weakness95,103; cases with antenatal onset or presentation in adulthood104–107 have been reported, but the molecular defect in these is not known. Clinical features associated with the histopathological appearance of MmD are markedly heterogeneous, and at least four different subgroups have been recognized and are gradually being resolved molecularly (see later discussion).
The most instantly recognizable classic phenotype of MmD95,103,108 usually manifests in infancy with hypotonia and is characterized by spinal rigidity, scoliosis, and early respiratory impairment. There is predominant truncal and proximal weakness, often in association with wasting pronounced in the shoulder girdle and the hip adductors (“bracket-like thighs”). There may be mild facial weakness and a high-pitched nasal voice. Severe scoliosis and respiratory failure have usually evolved by the early teens.95,103,109 Respiratory impairment is often disproportionate to the overall degree of muscle weakness and has to be anticipated even in ambulant patients. Most of these cases are secondary to mutations in the SEPN1 gene (discussed later).
In a subset of patients with a similar phenotype, extraocular muscle involvement pronounced on abduction and upward gaze may evolve over time.94,95,110 With the exception of the most severely affected neonatal cases,51 respiratory impairment is usually milder than in the classic form and may improve with time. Mutations in the RYR1 gene have been identified in these patients (discussed later).
Another group has a milder phenotype similar to CCD and characterized by predominant hip girdle weakness with relative sparing of respiratory and bulbar muscles.37 A common complaint is exercise-induced myalgia. In male patients, cryptorchism may be an additional feature (personal observation). Patients in this group may also demonstrate muscle imaging findings similar to those observed in classic CCD caused by mutations in the RYR1 C-terminal portion.37,87,110 In some patients, there is additional marked distal weakness and wasting, predominantly affecting the hands.37,87 The observation of extraocular muscle involvement evolving over time in this group is suggestive of a clinical continuum between the latter groups rather than distinct clinical entities.110 Mutations in the RYR1 gene also account for this group of disorders.
A severe form with antenatal onset, generalized arthrogryposis, dysmorphic features, and moderate respiratory impairment has been described in only a few patients.103
Congenital cardiac defects—namely, mitral valve prolapse—have been occasionally reported.5,37 Secondary right ventricular impairment is common in the classic phenotype with untreated respiratory impairment but not in other clinical subgroups.37 A primary cardiomyopathy has been reported in some cases with minicores on muscle biopsy specimens,55,106,111,112 but these were genetically unresolved, and additional desmin accumulation in muscle and dominant inheritance were suggestive of a pathogenic mechanism distinct from that in other families (see later discussion).
Malignant hyperthermia has been reported occasionally113,114 and has to be anticipated in the anesthetic management of these patients. Minicores have been noted in muscle biopsy specimens from a few families with mutations in the RYR1 gene and MHS but no other clinical features of a congenital myopathy.115,116
Minicores have been reported in the multiple pterygium syndrome117 and in two siblings with mental retardation and dysmorphic features similar to the King-Denborough syndrome118; the molecular basis of these associations is currently unclear.
The clinical course is usually static or only slowly progressive37,119 but depends largely on the degree of cardiorespiratory impairment.
Genetics
Investigation of SEPN1 on chromosome 1p36 as a candidate for causing MmD was prompted by the considerable clinical and histopathological overlap between the classic phenotype of MmD and congenital muscular dystrophy with rigidity of the spine, previously attributed to SEPN1 mutations.120 SEPN1 involvement in the classic phenotype of MmD was suggested both by linkage data and by direct mutational analysis in approximately 50% of patients with these clinical features.108 More than 30, mainly truncating SEPN1 mutations associated with a congenital myopathy phenotype have been identified to date. Mutations in the SEPN1 gene were also identified in cases with unusual pathological structures resembling Mallory bodies (Mallory body myopathy), further expanding the pathological spectrum of the condition.121 Homozygous mutations are unexpectedly common even in nonconsanguineous families, which reflects the presence of a few founder mutations in different European populations. The precise function of selenoprotein N, a glycoprotein localized in the endoplasmic reticulum, is unclear, but a structural motif similar to those observed in calcium-binding proteins120 is suggestive of possible involvement in calcium homeostasis in muscle. Although selenoprotein N is expressed in all adult tissues, prominent expression in fetal muscle precursor cells is suggestive of a potential role in myogenesis.
Involvement of the RYR1 gene as a cause of multi-minicores was to some extent unexpected, in view of differences in core structure and mode of inheritance between recessively inherited MmD and CCD caused by dominant mutations in the same gene. Attempting to classify individual cases as MmD or CCD may be difficult because there is a histopathological and clinical continuum between the two conditions. Although the histopathologic appearance of MmD appears to be more closely associated with recessively inherited RYR1 mutations, dominant RYR1 mutations occasionally give rise to minicores on muscle biopsy specimens10 and may have accounted for a proportion of MmD pedigrees with autosomal dominant inheritance reported before the molecular area. Also, the histopathological appearance of MmD caused by recessive RYR1 mutations may evolve into the classic picture of CCD over long periods of follow-up.87 A particular feature of RYR1-related recessive cases with minicores is extraocular involvement, not present in dominant CCD. MmD with external ophthalmoplegia in an isolated case from a consanguineous Tunisian family was attributed to a homozygous RYR1 mutation, introducing a cryptic splice site in intron 101,51 carried by asymptomatic parents. RYR1 involvement was also suggested by linkage evidence in four additional families with a similar phenotype,110 including the original family reported by Swash and Schwartz.94 RYR1 involvement in the moderate form of MmD with hand involvement was suggested by considerable clinical overlap with CCD and an identical pattern of selective involvement on muscle imaging.37,87 In a consanguineous Algerian family and a consanguineous British family, homozygous RYR1 mutations were subsequently identified, and RYR1 involvement has been suggested by linkage evidence in other families. The RYR1 gene is also a likely candidate for causing the severe form of MmD with neonatal onset and arthrogryposis, in view of phenotypic overlap with the form of CCD with fetal akinesia sequence.13
There is evidence for further genetic heterogeneity in MmD, because only one half of all cases with the classic phenotype have linkage or mutational evidence of SEPN1 involvement.108 Also, although a proportion of MmD in pedigrees with autosomal dominant inheritance are likely to be caused by dominant RYR1 mutations, unusual clinical features such as a primary cardiomyopathy55,111 are suggestive of a different genetic background in other families with this unusual mode of transmission. Specimens from patients with dominantly inherited desmin myopathy may demonstrate minicores in addition to inclusions on muscle biopsy. This observation and the desmin accumulation reported in some patients with MmD and cardiomyopathy111 suggest that the desmin gene is a possible candidate for causing this subgroup disorder. Also, a cardiomyopathy associated with cores on muscle biopsy has been described as part of the expanding clinical spectrum associated with dominant mutations in the ACTA1 gene.20
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


