, Jean Paul G. Vonsattel2, Helmut Heinsen3, 4 and Horst-Werner Korf5
(1)
Dr. Senckenbergisches Chronomedizinisches Institut, Goethe University Frankfurt, Frankfurt, Germany
(2)
Medical Center Neurological Institute, Columbia University, New York, NY, USA
(3)
Division Psychiatic Clinic Morphological Brain Research Unit, Julius Maximilians University Würzburg, Würzburg, Germany
(4)
University of Sao Paulo Medical School, Sao Paulo, Brazil
(5)
Dr. Senckenbergisches Chronomedizinisches Institut, Goethe University, Frankfurt, Frankfurt, Germany
9.1 The Disease Protein Huntingtin
Identified in 1993 and located on chromosome 4p16.3, the human HD gene (also called IT15) contains 67 exons and spans more than 200 kb. It harbors meiotically unstable CAG trinucleotide or polyglutamine repeats (cytosine, adenine, guanine) in its exon 1, which encode the very large 350 kDa huntingtin protein (Htt). Htt consists of about 3100 amino acids and undergoes extensive posttranslational modifications (Fig. 9.1) (Andrew et al. 1993; Atkin and Paulson 2014; Borrell-Pagès et al. 2006; Brundin et al. 2010; Cattaneo et al. 2005; Davies et al. 2007; Duyao et al. 1993; Finkbeiner and Mitra 2008; Imarisio et al. 2008; Labbadia and Morimoto 2013; Li and Conforti 2013; Li and Li 2011; Margolis and Ross 2003; Myers et al. 1991; Ortega et al. 2007; Paul 2008; Renner and Melki 2014; Schulte and Littleton 2011; The Huntington’s disease Collaborative Research Group 1993; Vonsattel 2008; Walker 2007a, b).
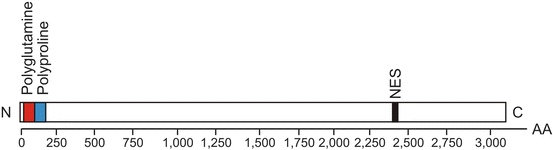
Fig. 9.1
The gene of the polyglutamine disorder Huntington’s disease (HD). The human Huntington (HD) gene (also called IT15 gene) contains 67 exons and spans more than 200 kb. In its N-terminus (N) it harbors meiotically unstable CAG trinucleotide repeats (red) which encode for the large polyglutamine protein huntingtin (Htt). In affected Huntington’s disease (HD) patients and asymptomatic gene carriers, the CAG trinucleotide or polyglutamine repeats at the N-terminus (N) are pathologically expanded and give rise to an elongated polyglutamine tract in mutant Htt that confers the tendency of Htt to form insoluble intraneuronal inclusions. Depending on the length of the polyglutamine tract, Htt consists of about 3100 amino acids. Although the elongated polyglutamine tract at the N-terminus (N) of mutant Htt in higher vertebrates is flanked by a polyproline domain (blue) which is thought to help to maintain solubility of this protein, elongations of the polyglutamine tract in mutant Htt result in conformational changes, misfolding, and reduced solubility of Htt. The C-terminus (C) of Htt has an active nuclear export signal (NES) (black) which serves shuttling transcription factors between the nucleus and the cytoplasm of nerve cells (Modified according to Cattaneo et al. (2005) (Figure 1, page 922); with kind permission from Nature Publishing Group). Abbreviations: AA amino acid, C C-terminus, Htt huntingtin, N N-terminus, NES nuclear export signal
In symptomatic HD patients and asymptomatic gene carriers, the CAG trinucleotide and the polyglutamine repeats are pathologically expanded and give rise to an elongated polyglutamine tract at the N-terminus of mutant Htt that confers the tendency to Htt to form intraneuronal inclusions (Fig. 9.1) (Andrew et al. 1993; Atkin and Paulson 2014; Borrell-Pagès et al. 2006; Cattaneo et al. 2005; Duyao et al. 1993; Finkbeiner and Mitra 2008; Gunawardena and Goldstein 2005; Imarisio et al. 2008; Labbadia and Morimoto 2013; Li and Conforti 2013; Li and Li 2011; Margolis and Ross 2003; Myers et al. 1991; Ortega et al. 2007; Paul 2008; Renner and Melki 2014; Schulte and Littleton 2011; The Huntington’s disease Collaborative Research Group 1993; Vonsattel 2008; Walker 2007a, b; Wooten et al. 2006). The normal physiological CAG repeat sequences comprise 6–35 CAG triplets, whereby sequences of 28 and more already behave unstable during meiosis and may be prone to potentially symptomatic mutations. CAG repeat sequences longer than 35 are considered pathologically expanded, 36–40 CAG repeats lead to an incomplete penetrance, and expansions of 41 or more CAG repeats lead to the fully developed clinical picture of HD (Andrew et al. 1993; Atkin and Paulson 2014; Borrell-Pagès et al. 2006; Brundin et al. 2010; Cattaneo et al. 2005; Duyao et al. 1993; Finkbeiner and Mitra 2008; Imarisio et al. 2008; Labbadia and Morimoto 2013; Margolis and Ross 2003; Myers et al. 1991; Ortega et al. 2007; Schulte and Littleton 2011; The Huntington’s disease Collaborative Research Group 1993; Walker 2007a, b).
The translated wild–type polyglutamine protein Htt with its polymorphic stretch of between 6 and 35 glutamines in its N-terminal domain represents a large 350-kDa protein, which may traffic between the nucleus and the cytoplasm of nerve cells. Htt shows a very high degree of conservation among vertebrates, but no homologies with other proteins. Htt is expressed ubiquitously throughout the human body with its highest levels in the brain and testes, is found in all neurons and all glial cells of the brain, and has its main brain expression sites in the cerebral neocortex, cerebellar cortex, striatum, and hippocampus (Borrell-Pagès et al. 2006; Cattaneo et al. 2005; Finkbeiner and Mitra 2008; Imarisio et al. 2008; Labbadia and Morimoto 2013; Li and Li 2011; Millecamps and Julien 2013; Ortega et al. 2007; Schulte and Littleton 2011; Vonsattel 2008; Walker 2007a, b).
The subcellular neuronal localization of the polyglutamine protein Htt is complex and dynamic. Htt may change its conformation depending on its compartmental localization in nerve cells, is mainly found in the cytoplasm of nerve cells, and co-localizes with many organelles including the nucleus, endoplasmic reticulum, Golgi complex, mitochondria, and endosomes. Htt is also observed in axonal processes, at synapses, as well as in association with microtubules, synaptic and autophagic vesicles, caveolae, and synaptosomes (Atkin and Paulson 2014; Borrell-Pagès et al. 2006; Cattaneo et al. 2005; Davies et al. 2007; Imarisio et al. 2008; Labbadia and Morimoto 2013; Li and Conforti 2013; Li and Li 2011; Millecamps and Julien 2013; Pandey et al. 2010; Schulte and Littleton 2011).
Htt interacts with many proteins that regulate endocytosis. Owing to its association with the endocytosis proteins clathrin and dynamin, as well as with endocytic organelle trafficking proteins, Htt is also involved in the short-range transport along the actin cytoskeleton as a component of the endocytotic pathway and as such is thought to allow Htt-associated endocytic vesicles to move along actin filaments.
The C-terminus of Htt has an active nuclear export signal (NES), while its N-terminus domain forms an amphipathic alpha-helical membrane-binding domain that reversibly mediates the association of Htt with the endoplasmic reticulum, endosomes, and autophagic vesicles (Fig. 9.1). Htt has antiapoptotic activity and a large number of protein–protein interaction domains. Owing to these domains it can interact with over 200 other cellular proteins including those involved in gene expression, intracellular signaling, intracellular transport of signaling molecules and trophic factors, as well as metabolism. Htt also appears to be involved in various functions in the neuronal cytoplasm and nucleus, binds to and interacts with various transcription factors, and may regulate transcription by shuttling transcription factors between the nucleus and the cytoplasm using its NES and by interacting with spliceosome-related proteins (Atkin and Paulson 2014; Borrell-Pagès et al. 2006; Davies et al. 2007; Imarisio et al. 2008; Millecamps and Julien 2013; Schulte and Littleton 2011).
The Htt protein also acts as a scaffold that links transport cargos with the molecular motor proteins of the axonal transport and additionally regulates factors that coordinate trafficking and transport of cellular material along and between actin and microtubule cytoskeletons in a bidirectional manner over both short and long distances. One of the main proteins interacting with Htt is the huntingtin-associated protein HAP1, which helps to mediate the interaction between Htt and the molecular motor proteins of anterograde and retrograde axonal transport mechanisms (see Sect. 8.1) (Fig. 8.2).
Moreover, evidence is also available that Htt is involved in the sorting mechanisms of many proteins at the Golgi region (Atkin and Paulson 2014; Borrell-Pagès et al. 2006; Cattaneo et al. 2005; Davies et al. 2007; De Vos et al. 2008; Gunawardena and Goldstein 2005; Imarisio et al. 2008; Labbadia and Morimoto 2013; Li and Conforti 2013; Li and Li 2011; Millecamps and Julien 2013; Ortega et al. 2007; Schulte and Littleton 2011). Finally, Htt is associated with the antero– and retrograde movement of mitochondria along axons of nerve cells (Figs. 8.1 and 8.2).
Along with that of other cargos, the anterograde and retrograde trafficking of mitochondria in axons may be impeded and physically blocked by intraneuronal protein aggregates in HD. This early disruption of mitochondrial trafficking in nerve cells leads to a reduced deposition of mitochondria at sites with a high local energy demand such as synapses; reduces the number of mitochondria at essential subcellular locations and their sufficient, timely, and specific local energy supply; and ultimately results in an impaired synthesis of ATP. Since both axonal trafficking and mitochondrial dysfunction may occur in HD, the links between these two processes during disease pathogenesis may be important for considerations of therapeutic intervention (see Sect. 8.1) (Fig. 8.2) (Borrell-Pagès et al. 2006; Davies et al. 2007; De Vos et al. 2008; Gil and Rego 2008; Gunawardena and Goldstein 2005; Imarisio et al. 2008; Labbadia and Morimoto 2013; Li and Conforti 2013; Li and Li 2011; Millecamps and Julien 2013; Oliveira 2010; Ortega et al. 2007; Pandey et al. 2010; Rüb et al. 2013a, 2014a; Schulte and Littleton 2011; Walker 2007a, b).
Although the elongated polyglutamine tract of mutant Htt in higher vertebrates is flanked by a polyproline domain which is thought to help to maintain solubility of this protein (Fig. 9.1), elongations of the polyglutamine tract in mutant Htt result in conformational changes, misfolding, and a reduction of the solubility of this protein. All these processes most likely overload the defense mechanisms of affected nerve cells (i.e., molecular chaperones, UPS) necessary for cellular homeostasis of protein recycling and energetics and eventually lead to the formation of insoluble ubiquitinated neuronal protein aggregations (Atkin and Paulson 2014; Borrell-Pagès et al. 2006; Brundin et al. 2010; Cattaneo et al. 2005; Davies et al. 2007; Finkbeiner and Mitra 2008; Imarisio et al. 2008; Jucker and Walker 2011; Labbadia and Morimoto 2013; Ortega et al. 2007; Schulte and Littleton 2011; Walker 2007a).
To date, the molecular basis of the formation of the abnormally ubiquitinated neuronal aggregations is only fragmentarily understood. However, it is well known that these aggregations that evolve during the course of the polyglutamine disease HD contain, along with the small degradation-related 8 kDa ubiquitin protein, also highly ordered amyloid fibers with high β-sheet structure content (Fig. 9.1) and numerous other proteins, including factors important for transcription and protein quality control (e.g., proteasome subunits and molecular chaperones) (Atkin and Paulson 2014; Becher et al. 1998; Borrell-Pagès et al. 2006; Finkbeiner and Mitra 2008; Gunawardena and Goldstein 2005; Gutekunst et al. 1999; Imarisio et al. 2008; Labbadia and Morimoto 2013; Leak 2014; Li and Li 2011; Maat-Schieman et al. 1999; Ortega et al. 2007; Paul 2008; Schulte and Littleton 2011; Sieradzan et al. 1999; Walker 2007a, b; Wooten et al. 2006). Owing to the striking tendency of mutant Htt to form insoluble neuronal inclusions or aggregations, the pathological process of HD, like that of other known human proteinopathies or polyglutamine diseases, is associated with the occurrence of insoluble neuronal aggregations which represent the most striking immunocytochemical hallmarks of the HD-associated brain pathology (Atkin and Paulson 2014; Borrell-Pagès et al. 2006; Finkbeiner and Mitra 2008; Imarisio et al. 2008; Labbadia and Morimoto 2013; Li and Conforti 2013; Li and Li 2011; Maat-Schieman et al. 2007; Schulte and Littleton 2011; Walker 2007a, b; Wooten et al. 2006).
9.2 Types, Composition, and Pathogenetic Relevance of Neuronal Protein Aggregations in Huntington’s Disease (HD)
In higher vertebrates the elongated polyglutamine tract of mutant Htt is flanked by a polyproline domain which is thought to help to maintain solubility of this protein (Fig. 9.1). The pathological elongations of the polyglutamine tract in mutant Htt result in conformational changes, misfolding, and a reduction of the solubility of the Htt protein which most likely overload the defense mechanisms of the affected nerve cells (i.e., molecular chaperones, ubiquitin-proteasome pathway) necessary for cellular homeostasis of protein recycling and energetics and eventually lead to the formation of insoluble ubiquitinated neuronal protein aggregations (Atkin and Paulson 2014; Becher et al. 1998; Borrell-Pagès et al. 2006; Finkbeiner and Mitra 2008; Gunawardena and Goldstein 2005; Imarisio et al. 2008; Labbadia and Morimoto 2013; Leak 2014; Li and Li 2011; Maat-Schieman et al. 1999; Ortega et al. 2007; Schulte and Littleton 2011; Sieradzan et al. 1999; Walker 2007a, b; Wooten et al. 2006). Although, to date, the molecular basis of their formation is only fragmentarily understood, it is well known that the abnormal neuronal aggregations that evolve during the course of HD along with the small 76 amino acid protein ubiquitin also contain highly ordered amyloid fibrils with high β-sheet content and numerous other proteins, including factors important for transcription and components of the neuronal protein quality machinery (e.g., proteasome subunits and molecular chaperones) (Atkin and Paulson 2014; Becher et al. 1998; Borrell-Pagès et al. 2006; Davies et al. 2007; DiFiglia et al. 1997; Finkbeiner and Mitra 2008; Gunawardena and Goldstein 2005; Gutekunst et al. 1999; Imarisio et al. 2008; Labbadia and Morimoto 2013; Li and Li 2011; Maat-Schieman et al. 1999; Paul 2008; Schulte and Littleton 2011; Sieradzan et al. 1999; Walker 2007a, b; Wooten et al. 2006). Owing to the striking tendency of the mutated form of the polyglutamine protein Htt to form insoluble neuronal inclusions or aggregations, the pathological process of HD, like that of other known human proteinopathies or polyglutamine diseases, is associated with the occurrence of insoluble neuronal aggregations that represent the most prominent concomitant immunocytochemical hallmarks of the HD-associated brain pathology (Atkin and Paulson 2014; Becher et al. 1998; Borrell-Pagès et al. 2006; DiFiglia et al. 1997; Finkbeiner and Mitra 2008; Gil and Rego 2008; Gourfinkel-An et al. 1998; Gunawardena and Goldstein 2005; Gutekunst et al. 1999; Imarisio et al. 2008; Kuemmerle et al. 1999; Labbadia and Morimoto 2013; Li and Conforti 2013; Li and Li 2011; Maat-Schieman et al. 1999, 2007; Ortega et al. 2007; Paul 2008; Sapp et al. 1999; Schulte and Littleton 2011; Sieradzan et al. 1999; Valera et al. 2005; Walker 2007a, b).
Aggregations of Htt in the form of neuronal intranuclear inclusions (NI) were among the first types of neuronal aggregation pathologies described in HD, are well known as accompanying neuropathological feature of HD since more than two decades, and until recently were most frequently and nearly exclusively identified and described in the degenerated neostriatum and cerebral neo- and allocortex (Fig. 9.2) (Atkin and Paulson 2014; Becher et al. 1998; Borrell-Pagès et al. 2006; DiFiglia et al. 1997; Finkbeiner and Mitra 2008; Gil and Rego 2008; Gourfinkel-An et al. 1998; Gunawardena and Goldstein 2005; Gutekunst et al. 1999; Imarisio et al. 2008; Kuemmerle et al. 1999; Labbadia and Morimoto 2013; Li and Conforti 2013; Li and Li 2011; Maat-Schieman et al. 1999, 2007; Ortega et al. 2007; Rüb et al. 2014a; Sapp et al. 1999; Schulte and Littleton 2011; Sieradzan et al. 1999; Valera et al. 2005; Walker 2007b). Further biochemical studies showed that these subcortical and cortical NI along with the mutated disease protein Htt may also contain components of the cellular protein quality control machinery responsible for the handling, removing, and degrading of conformationally changed, misfolded, and polyubiquitin-tagged proteins (i.e., ubiquitin, proteasome subunits, molecular chaperones). Some previous neuropathological studies have demonstrated additional neuropil (i.e., neuritic or axonal) aggregates or swellings in select gray and white matter brain components of HD patients (i.e., neostriatum, cerebral neocortex, internal capsule, cerebral peduncle) (Atkin and Paulson 2014; Becher et al. 1998; Borrell-Pagès et al. 2006; Finkbeiner and Mitra 2008; Gil and Rego 2008; Gunawardena and Goldstein 2005; Gutekunst et al. 1999; Imarisio et al. 2008; Kuemmerle et al. 1999; Labbadia and Morimoto 2013; Li and Conforti 2013; Li and Li 2011; Maat-Schieman et al. 2007; Ortega et al. 2007; Paul 2008; Sapp et al. 1999; Schulte and Littleton 2011; Sieradzan et al. 1999; Valera et al. 2005; Walker 2007b; Wooten et al. 2006).
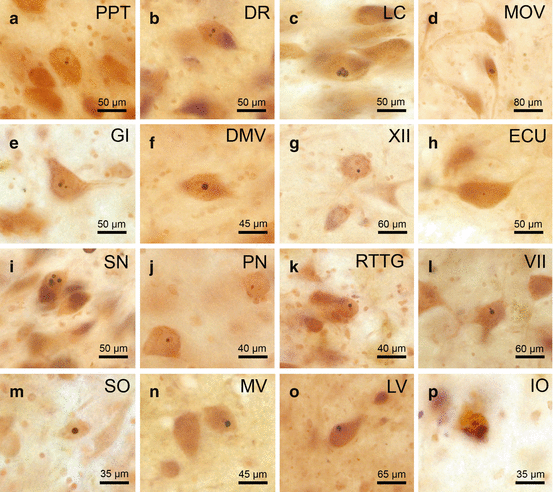
Fig. 9.2
Neuronal intranuclear inclusions in brainstem nuclei of Huntington’s disease (HD) patients immunoreactive for the ubiquitin-interacting and proteasomal shuttle protein p62. p62 immunoreactive neuronal intranuclear inclusions (NI) in well–preserved brainstem nuclei in Huntington’s disease (HD): (a) pedunculopontine nucleus (PPT), (b) dorsal raphe nucleus (DR), (c) locus coeruleus (LC), (d) motor trigeminal nucleus (MOV), (e) gigantocellular reticular nucleus (GI), (f) dorsal motor vagal nucleus (DMV), (g) hypoglossal nucleus (XII), (h) external cuneate nucleus (ECU). p62 immunoreactive NI in degenerated brainstem nuclei in HD: (i) substantia nigra (SN), (j) pontine nuclei (PN), (k) reticulotegmental nucleus of the pons (RTTG), (l) facial nucleus (VII), (m) superior olive (SO), (n) medial vestibular nucleus (MV), (o) lateral vestibular nucleus (LV), (p) inferior olive (IO) (a, b, f, l, n, o HD patient with Vonsattel grade 4 of neostriatal atrophy; 4; c, g, h, m HD patient with Vonsattel grade of neostriatal atrophy; d, i HD patient with Vonsattel grade 4 of neostriatal atrophy; e, j, k, p HD patient with Vonsattel grade 3 of neostriatal atrophy) (a–p: p62 immunohistochemistry, counterstaining with aldehyde-fuchsin Darrow red, 100 μm polyethylene glycol sections) (Reprinted from Rüb et al. (2014a), (Figure 4, page 254); with kind permission from John Wiley and Sons)
Recent systematic immunocytochemical studies of the brainstem and cerebellum of HD patients confirmed (1) that NI by no means represent the sole type of neuronal protein aggregations that develop during HD, (2) that NI are not confined to the classical brain regions known to undergo neurodegeneration in HD (i.e., neostriatum, cerebral cortex), and (3) that the neuronal protein aggregation pathology is more widespread in the brains of HD patients than hitherto thought (Figs. 9.2, 9.3, 9.4, 9.5, and 9.6) (Rüb et al. 2013a, 2014a). According to these studies (1) immunoreactive compact and granular neuronal protein aggregations may also occur in the cytoplasm of cerebellar nerve cells, (2) brainstem NI are present in both degenerated and spared nuclei of HD patients, and (3) the occurrence of intra–axonal inclusions in fiber tracts is a common and widespread feature of the cerebellar and brainstem pathology of HD patients (Figs. 9.2, 9.3, 9.4, 9.5, and 9.6) (Rüb et al. 2013a, 2014a).

Fig. 9.3
 Pathological Nerve Cell Alterations in Huntington’s Disease (HD) and Their Possible Role for the Demise of Nerve Cells
Conclusions and Outlook
Pathological Nerve Cell Alterations in Huntington’s Disease (HD) and Their Possible Role for the Demise of Nerve Cells
Conclusions and Outlook
 Elucidation of the Role of the Premotor Oculomotor Brainstem Nuclei in the Pathogenesis of Oculomotor Dysfunctions in Huntington’s Disease (HD)
Elucidation of the Role of the Premotor Oculomotor Brainstem Nuclei in the Pathogenesis of Oculomotor Dysfunctions in Huntington’s Disease (HD)
 Widespread Brainstem Neurodegeneration in Huntington’s Disease (HD)
Widespread Brainstem Neurodegeneration in Huntington’s Disease (HD)
 Intraneuronal Transport and Defense Mechanisms with Possible Pathogenetic Relevance in Huntington’s Disease (HD)
Intraneuronal Transport and Defense Mechanisms with Possible Pathogenetic Relevance in Huntington’s Disease (HD)
 Degeneration of Select Motor and Limbic Nuclei of the Thalamus in Huntington’s Disease (HD)
Degeneration of Select Motor and Limbic Nuclei of the Thalamus in Huntington’s Disease (HD)




Axonal inclusions in brainstem fiber tracts in Huntington’s disease (HD) immunoreactive for the ubiquitin-interacting and proteasomal shuttle protein p62. p62 immunoreactive axonal inclusions in Huntington’s disease (HD): (a) oculomotor nerve (III), (b) trochlear nerve (IV), (c) pontocerebellar fibers (PCF), (d) PCF, (e) trigeminal nerve (V), (f) facial nerve (VII), (G) trapezoid body (TZ), (h) vestibulocochlear nerve (VIII), (i) inferior cerebellar peduncle (ICP), (j) intermediate reticular zone (IRZ), (k) olivocerebellar fibers (OCF), (l) hypoglossal nerve (XII), (m) hypoglossal nerve (XII), (n) cuneate fascicle (ECU), (o) solitary tract (SOL), (p) dorsal spinocerebellar tract (DST). (a–p p62/AT270 double immunohistochemistry, 100 μm PEG sections) (a, e, k, n HD patient with Vonsattel grade 4 of neostriatal atrophy; b, l, p HD patient with Vonsattel grade 2 of neostriatal atrophy; c, d, f, g, h HD patient with Vonsattel grade 3 of neostriatal atrophy; i HD patient with Vonsattel grade 3 of neostriatal atrophy; j, o HD patient with Vonsattel grade 3 of neostriatal atrophy: 3; m HD patient with Vonsattel grade 4 of neostriatal atrophy) (Reprinted from Rüb et al. (2014a), (Figure 5, page 255); with kind permission from John Wiley and Sons)
Related posts:
Pathological Nerve Cell Alterations in Huntington’s Disease (HD) and Their Possible Role for the Demise of Nerve Cells
Conclusions and Outlook
Elucidation of the Role of the Premotor Oculomotor Brainstem Nuclei in the Pathogenesis of Oculomotor Dysfunctions in Huntington’s Disease (HD)
Widespread Brainstem Neurodegeneration in Huntington’s Disease (HD)
Intraneuronal Transport and Defense Mechanisms with Possible Pathogenetic Relevance in Huntington’s Disease (HD)
Degeneration of Select Motor and Limbic Nuclei of the Thalamus in Huntington’s Disease (HD)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
