Fig. 6.1
Hypertensive artery remodeling. This figure depicts the effects of hypertension on cerebral artery structure, a normotensive artery is shown in the left, and two forms of hypertensive artery remodeling are depicted on the right [22, 38, 49, 63–75, 78]. During inward hypertrophic artery remodeling the lumen diameter of the artery is reduced, and the wall thickness, wall-to-lumen ratio, and cross-sectional wall area are increased. The increase in the wall area (shown in red) results from enhanced smooth muscle cell proliferation or growth. During eutrophic artery remodeling the lumen diameter is reduced and the wall thickness and wall-to-lumen ratio are both increased but the cross-sectional wall area of the artery does not increase. The text box on the right of the figure described some of the proposed mechanisms to prevent or reverse cerebral artery remodeling [64, 73, 79–89, 91, 92, 101, 106–109, 132, 135]
In cerebral arteries the increased wall thickness is generally associated with smaller lumen diameter [51, 60], and an elevated wall-to-lumen ratio. An increased wall-to-lumen ratio is a good predictor of end-organ damage [61] that can result in stroke [62]. Although it is clear that the wall-to-lumen ratio is increased in cerebral arteries from hypertensive rats the effects of hypertension on the artery wall cross-sectional area have been more difficult to define. This may be partly a function of the methodology used to study these arteries. In pressure myograph and cranial window studies wall cross-sectional area is a derived variable; this causes compounding of experimental errors and leads to a situation where statistical significance is difficult to achieve. Histology or immunostaining may provide more definitive results for wall cross-sectional area. It should be noted that processing samples for histology or immunostaining can cause tissue shrinkage and this may make it difficult to identify subtle changes in artery structure and remodeling.
In rat models of essential hypertension, such as the SHR and SHRSP, the large arteries like the middle cerebral arteries [63, 64] and the pial arteries [49] have smaller lumens and thicker walls. This type of change in middle cerebral artery structure is also found in several models of secondary hypertension such as obesity-induced hypertension [65, 66], mineralocorticoid receptor-dependent hypertension [67, 68], and N-nitro-l-arginine methyl ester (l-NAME)-dependent hypertension [69]. DOCA-salt hypertensive rats exhibit a reduction in the lumen diameter of their pial arteries [38], but the larger cerebral arteries have not been studied. Other models of secondary hypertension do not behave in the same way; for example, there is no difference in the lumen diameter of the pial arteries from rats with renal hypertension compared to control rats, but the wall thickness is increased. Although it was not reported, this combination of changes should increase the wall-to-lumen ratio [70]. These studies suggest that elevations in blood pressure alone are not the only stimulus for the inward artery remodeling observed in many hypertensive models. The blood pressure dependency of the cerebral artery remodeling process has been a fruitful research area.
Pial artery remodeling develops in SHRSP as they age and their blood pressure increases. Young (3–4 months old) SHRSP do not exhibit pial artery remodeling, but in aged SHRSP (10–12 months old) the pial arteries exhibit a clear inward remodeling when compared to arteries from normotensive Wistar Kyoto (WKY) rats [49, 71]. However, both young and aged SHRSP have marked hypertension [72], suggesting that the pial arteries in the younger rats maintain a normal structure in the face of elevated blood pressure. The pial arteries are downstream from the larger cerebral arteries like the middle cerebral artery, and it seems that this artery undergoes remodeling earlier in the genesis of hypertension. Middle cerebral arteries from 12-week-old SHRSP have smaller lumens and an increased wall-to-lumen ratio compared to arteries from WKY rats [63, 64, 73]. These studies suggest that different segments of the cerebrovascular tree remodel at different rates in response to hypertension. It appears that artery remodeling, and artery rarefaction, progresses from the large arteries to the small arteries over time. This temporal progression of the artery remodeling process is in keeping with concept that vascular resistance increases in the smaller vessels in the sustained phase of the hypertension [22]. Studies in female SHR have shown that the penetrating arterioles also undergo artery remodeling. The arterioles from 18-week-old SHR have a smaller lumen diameter than the same arteries from WKY rats; the authors did not describe effects of hypertension on wall thickness in this study [74]. At present it is unclear if the timeframe of the remodeling in the penetrating arterioles differs from the pial arteries they branch from.
A similar temporal development of artery remodeling has been observed in obese Zucker rats that develop hypertension as they age and obesity develops. Osmond et al. studied the middle cerebral arteries from young (6–7 weeks old) and adult (14–16 weeks old) obese Zucker rats [66]. All the rats were obese, but only the adult rats where hypertensive. The young rats showed no signs of middle cerebral artery remodeling, but the arteries from the older obese hypertensive rats underwent an inward remodeling that included a reduction in the lumen and outer diameter and a small increase in wall thickness [66]. In a follow-up study, Osmond et al. investigated the effects of blood pressure lowering on artery remodeling in obese Zucker rats. Hydrochlorothiazide, an antihypertensive drug, prevented the hypertension and the middle cerebral artery remodeling observed in the obese Zucker rats. These effects occurred independently of body weight, insulin, and triglyceride or cholesterol levels. These studies strongly suggest that hypertension is an important determinant of cerebral artery remodeling in the obese Zucker rats [75].
Interestingly, cerebral artery remodeling in SHRSP is accompanied by changes in the organization of the smooth muscle cells in the artery wall. In arteries from normotensive rats the smooth muscle cells are arranged circularly around the artery wall, and the long axis of the muscle cell runs at a 90° angle to the direction of blood flow. In basilar arteries from SHRSP this organization is lost in some discrete regions of the wall where several smooth muscle cells were orientated such that their long axis is no longer perpendicular to the direction of blood flow [76, 77]. The thickness of the adventitia was also reduced in the areas were the smooth muscle cells were disorganized. The authors propose that the disorganized regions are weak areas in the artery that could increase the risk of hemorrhage in SHRSP [76]. Areas of cell disorganization could account for the increased risk of hemorrhage observed in SHSRP; however, this would only be the case if the areas of disorganized smooth muscle cells occurred throughout the cerebral vasculature and not just in the basilar artery. The changes in the organization of smooth muscle cells were also associated with impaired constriction caused by smooth muscle cell depolarization, suggesting that the proper smooth muscle cell orientation is vital to maintain the constrictor ability of smooth muscle cells in the arterial wall. Similar studies have been conducted in rats with experimental l-NAME-induced hypertension. Artery remodeling was observed in the l-NAME hypertensive rats but there were no regions with disorganized smooth muscle cells [78]. The duration or magnitude of the hypertension may contribute to the disparity in the results obtained with SHRSP and l-NAME hypertensive rats. The latter were treated with l-NAME for 3 weeks, and would have been hypertensive for only a portion of that time, whereas the SHRSP were 14 weeks old when they were studied and so would have had significant hypertension for at least 6 weeks [72]. However it is also important to note that the changes in the cell orientation observed in the SHRSP could be a genetic trait peculiar to this strain of rats.
6 The Renin-Angiotensin-Aldosterone System and Artery Structure
There is no doubt that the hemodynamic effects of hypertension are an important determinant of artery remodeling. However, there is mounting evidence suggesting that several circulating factors, which are increased in hypertensive subjects, also play an important role in the remodeling process.
The search for a molecular mechanism for artery remodeling has led to an intense scrutiny of the renin-angiotensin-aldosterone system (RAAS). Studies utilizing angiotensin-converting enzyme (ACE) inhibitors [79, 80] or angiotensin receptor blockers [81, 82] have helped define the blood pressure dependency of the cerebral artery remodeling process. The effects of RAAS inhibition have been compared to the effects of β-blockers, which lower blood pressure by a RAAS-independent mechanism. The key finding from these studies is that blood pressure lowering alone does not improve pial [79, 82] or middle cerebral artery structure [83, 84] in SHR or SHRSP. The β-blockers reduced blood pressure but did not alter artery structure; they did however reduce plasma renin activity and angiotensin II levels [85]. Why β-blockers and ACE inhibitors have disparate effects on artery structure remains a mystery. At a simplistic level one would expect them to have similar effects because both reduced blood pressure and angiotensin II levels. Other studies have utilized combinations of low doses of ACE inhibitors (ramipril) and angiotensin receptor blockers (telmisartan). These studies showed that targeting the RAAS with these drugs together improved pial artery structure and function in young SHR. Given alone the same doses of each drug had no effect [86]. However, the combination therapy was not more effective than a higher dose of either drug used alone [81]. In all these studies the drugs that improved the artery structure also reduced the blood pressure. Several of these studies also assessed cerebral blood flow and found that when the passive lumen diameter of the cerebral arteries was increased the cerebral blood flow was also elevated [81, 86, 87]. This suggests that passive artery structure plays a role in the control of cerebral blood flow.
Two recent studies assessed the effects of start term inhibition of the RAAS on pial and middle cerebral artery structure. In these studies 4–5-month-old SHR and WKY rats were treated with candesartan or telmisartan for 10 days. Both drugs reduced the blood pressure in the SHR, but only telmisartan increased the resting lumen diameter of the pial arterioles. The authors propose that the beneficial effects of telmisartan occur through a peroxisome proliferator-activated receptor γ (PPARγ)-dependent mechanism [88]. The effects of direct PPARγ activation on artery remodeling will be described further later. Interestingly telmisartan had no effect on the structure of the middle cerebral arteries [89] in rats undergoing the same treatment regime. This differential effect of angiotensin receptor blockade on the middle cerebral and pial arteries is interesting, but at present unexplained.
The studies described above are supported by studies utilizing angiotensinogen knockout mice; these mice do not produce angiotensin II and therefore are good correlates for studies using ACE inhibitors and angiotensin receptor blockers. This study focused on the small collateral arteries that connect the anterior and middle cerebral arteries. The lumen diameter of these anastomoses was increased in the mice lacking the angiotensinogen gene compared to wild-type mice. There was no difference in the number of anastomoses present between the two strains. The absence of angiotensin II in these mice also caused a reduction in blood pressure, and it is possible that it had significant effects on the artery structure [90].
One of the issues with many of the studies of hypertension-associated artery remodeling is the age of the rats and the timing of the treatments. In many cases the drug treatments were given as the hypertension was developing, in this situation effective drugs are essentially preventing artery remodeling. Clinically, it is much more important to identify ways to improve artery structure and function after the development of the hypertension and artery remodeling. Of particular note is a study showing that ACE inhibitors effectively improved artery structure in adult SHR; these rats were already hypertensive and artery remodeling would be present. Rats were treated with captopril from 12 months of age, and the studies were conducted in 15-month-old rats. The captopril treatment reduced the wall thickness and the wall-to-lumen ratio; captopril also reduced the systemic blood pressure. This study suggests that the hypertension associated with pial artery remodeling is reversible contingent upon a chronic reduction in blood pressure [87].
Aldosterone is the last signaling molecule produced by the RAAS. Aldosterone activates the mineralocorticoid receptor, and this has detrimental effects on cerebral arteries. Mineralocorticoid receptor activation with DOCA or 11β-hydroxysteroid dehydrogenase inhibition causes marked middle cerebral artery remodeling that is associated with the development of mild hypertension (systolic pressure of approximately 160 mmHg). These treatments induced an inward hypertrophic remodeling and the changes in artery structure translated into a large area of cerebral infarct when ischemia was induced experimentally [67, 68]. Conversely, mineralocorticoid receptor antagonists have beneficial effects on the cerebral vasculature in SHSRP. Treatment of young SHRSP from 6 to 12 weeks of age with spironolactone prevented the development of inward artery remodeling, the spironolactone-treated rats had middle cerebral arteries with larger lumens and the wall-to-lumen ratio was reduced [64]. Spironolactone also effectively reversed middle cerebral artery remodeling in older SHRSP with established hypertension. In this study the spironolactone-treated rats had larger middle cerebral artery lumen diameters than the untreated hypertensive rats [91]. The mineralocorticoid receptor antagonists did not lower the blood pressure in either study suggesting that the prevention and reversal of mineralocorticoid receptor-mediated artery remodeling is not dependent on systemic arterial pressure. Mineralocorticoid receptor activation has also been implicated in hypertension-associated endothelial dysfunction in the cerebral vasculature. This effect of aldosterone in the basilar artery appears to be the result of increased oxidative stress [92].
Rats treated with ACE inhibitors and angiotensin receptor blockers continue to show beneficial effects of the drugs even after treatment withdrawal [93–95]. A recent study assessed if the beneficial effects of mineralocorticoid receptor antagonists on cerebral arteries were sustained after their withdrawal. Surprisingly, withdrawal of spironolactone after a prolonged treatment had marked detrimental effects on the middle cerebral arteries from SHRSP. The rats were treated from 6 to 12 weeks of age and studied at 18 weeks of age. The middle cerebral artery lumen diameter was smaller in the rats treated with spironolactone when compared to the untreated SHRSP. The reduction in lumen diameter occurred without a change in the wall thickness or wall-to-lumen ratio. Although the structural effects of spironolactone withdrawal were deemed to be detrimental, a marked improvement in middle cerebral artery endothelium-dependent dilation was observed after spironolactone treatment and withdrawal [96]; this may be an effort by the vasculature to compensate for the inward artery remodeling. Interestingly, unlike ACE inhibitors and angiotensin receptor blockers, the effects of mineralocorticoid receptor activation on the cerebral arteries occurred in a blood pressure-independent manner [64, 91, 96]. In the future it will be important to investigate whether angiotensin II and aldosterone act synergistically to exacerbate the artery remodeling process.
The molecular mechanisms responsible for the changes in artery structure that occur with RAAS activation have not been completely elucidated. But the RAAS activates intracellular signaling cascades that could stimulate artery remodeling. Both angiotensin II and aldosterone increase reactive oxygen species production by increasing NADPHoxidase expression and activity [97, 98]. Superoxide is the reactive oxygen species most commonly produced in this situation, and increased superoxide production has been linked to artery remodeling in peripheral resistance [99] and coronary arteries [100] from hypertensive rats. Superoxide also stimulates remodeling in cerebral arteries. Treatment of SHRSP with tempol, a superoxide dismutase mimetic, prevented the reduction in the middle cerebral artery lumen diameter that is normally observed as hypertension develops in the SHRSP [101].
All forms of artery remodeling require some breakdown and reorganization of the extracellular matrix. In eutrophic inward remodeling the extracellular matrix breaks down to allow for the smooth muscle cells to rearrange themselves around a smaller lumen. In hypertrophic remodeling the extracellular matrix breaks down to accommodate hypertrophied smooth muscle cells or cell proliferation. Therefore, it is reasonable to propose that the matrix metalloproteinase (MMP) family of enzymes is involved in the artery remodeling process [102]. MMPs are zinc-dependent proteases responsible for breakdown and rearrangement of extracellular matrix components. Importantly, MMP expression is modulated by RAAS activation. Mineralocorticoid receptor antagonism reduces MMP-13 mRNA expression in large cerebral arteries from SHRSP [91], and angiotensin II increases MMP-2 and MMP-9 levels in smooth muscle cells [103]. Inhibiting the actions of MMPs effectively reduces the development of hypertension-associated cerebral artery remodeling. Chronic treatment of young SHRSP (6-week-old) with the nonspecific MMP inhibitor, doxycycline, increased the lumen diameter and reduced the wall-to-lumen ratio of the middle cerebral artery. As is the case with spironolactone, doxycycline had no effect on blood pressure suggesting the prevention of artery remodeling occurs through a blood pressure-independent mechanism. The damage produced by cerebral ischemia was also reduced by doxycycline treatment, and pial artery blood perfusion post-stroke was increased [73]. Importantly, doxycycline was withdrawn several days prior to the induction of cerebral ischemia; thus the improvement in the outcome of cerebral ischemia is probably not a response to acute MMP inhibition at the time of the stroke. This is important because clinical studies suggest that tetracycline antibiotics, like doxycycline and minocycline, have acute beneficial effects post-stroke [104, 105], but these effects do not appear to be vascular in nature.
7 Non-RAAS-Dependent Mechanisms of Artery Remodeling
In recent years the search of mediators of artery remodeling has moved beyond the RAAS system. Several of these potential mediators of the remodeling process will be discussed here.
7.1 Peroxisome Proliferator-Activated Receptor γ and HMG-CoA Reductase
l-NAME hypertensive rats develop inward remodeling of the large cerebral arteries and rosiglitazone, a PPARγ activator, prevents this. Importantly, the authors delayed the rosiglitazone treatment until the hypertension, and presumably the artery remodeling, was fully developed in the l-NAME-treated rats. This suggests that PPARγ activation reverses cerebral artery remodeling. Rosiglitazone had no effect on blood pressure in the l-NAME-treated rats [106] suggesting that its effects of artery structure are blood pressure independent. Treatment of New Zealand hypertensive rats with pioglitazone, another PPARγ activator, reduced the basilar artery media thickness. However, in this model, pioglitazone reduced systemic blood pressure, making it impossible to dissect apart the effects of PPARγ activation and blood pressure lowering [107]. It is not clear why rosiglitazone and pioglitazone had different effects on blood pressure; it is possible that this is a result of the different models of hypertension being used in these studies.
HMG-CoA reductase inhibitors (statins) have also been proposed as potential drugs for artery remodeling. In New Zealand hypertensive rats simvastatin reduced the wall thickness and increased the lumen diameter of the basilar artery [108]. A similar effect was observed in 2K2C hypertensive rats [109]. In both models simvastatin caused a marked reduction in blood pressure; therefore, it is unclear if the prevention of artery remodeling is a direct effect of the statin therapy or a response to the lower blood pressure.
7.2 Chloride Channels
The artery wall area increases during hypertrophic remodeling. This can occur as a result of smooth muscle cell hypertrophy (an increase in cell volume) or hyperplasia (cell proliferation). Irrespective of the mechanism for the increase in wall area the cell volume of the smooth muscle cells must increase. Cell volume is primarily controlled by the gain or loss of ions (Na+, K+, and Cl−) or small organic osmolytes including amino acids, polyols, and methylamines [110]. Two Cl− channels are important regulators of cell volume and cerebral artery remodeling; they will be discussed later. Receptor and store-operated Ca2+ channels [111], transient receptor potential (TRP) channels [112], large-conductance Ca2+-activated K+ channels, and voltage-gated K+ channels [113] have also been implicated in the cerebral artery remodeling process. We elected to focus on Cl− channels because this is an area where several developments have been made recently.
The volume-regulated Cl− channel (ICl.vol) is an important regulator of artery remodeling; ICl.vol activation controls smooth muscle cell volume, proliferation, and apoptosis [114]. The identity of the ICl.vol channel has been elusive, but CIC-3, a voltage-gated Cl− channel, is a candidate in vascular smooth muscle cells [114, 115]. Blocking ICl.vol causes rat cerebral artery smooth muscle cells to hyperpolarize and this causes dilation [116]. This suggests that ICl.vol activation will cause constriction in response to smooth muscle cell depolarization. ICl.vol activity has also been linked to the smooth muscle cell hypertrophy observed in basilar arteries from 2K2C rats; this increases the medial area of the basilar arteries compared to arteries from control rats. Cultured smooth muscle cells from 2K2C rats show increased ICl.vol activity when placed in a hypotonic solution. This increase in ICl.vol activity in the 2K2C rats is blood pressure and tyrosine kinase dependent [117]. Simvastatin treatment in 2K2C rats reduces the ICl.vol activity and artery remodeling [109].
More recent studies have confirmed the link between ICl.vol, CIC-3, and artery remodeling. Using CIC-3 knock out mice (CIC-3−/−) Zheng et al. showed that artery remodeling in DOCA-salt hypertensive mice was diminished. However CIC-3−/− mice treated with DOCA-salt did not develop as marked hypertension as the wild-type DOCA-salt-treated mice. The authors controlled for this by treating wild-type DOCA-salt mice with propranolol to lower their blood pressure to a level similar to the CIC-3−/− DOCA-salt-treated mice. Propranolol had no effect on artery remodeling, suggesting that the reduced artery remodeling in the DOCA-salt-treated CIC-3−/− mice was not merely a function of impaired response to the hypertensive agent, but rather it is specific to the knockout of the CIC-3−/− channel [118]. The same group of researchers has also shown that CIC-3 is involved in the development of angiotensin II-mediated artery remodeling. This study also linked the CIC-3 to integrin β3 and Src kinase [119]. This group of studies has come the closest to defining a molecular mechanism through which changes in blood pressure can drive the smooth muscle cell proliferation, apoptosis, and migration required for the genesis of artery remodeling. CIC-3 may play a key role in hypertension-associated artery remodeling because it is activated by angiotensin II, endothelin-1, and reactive oxygen species, and all these factors are increased in hypertension [120].
Changes in intracellular calcium may also be responsible for the ICl.vol-mediated artery remodeling. ICl.vol activation causes smooth muscle depolarization, and that will cause the voltage-gated Ca2+ channels to open. The resulting increase in intracellular calcium can activate smooth muscle cell proliferation [121, 122]; this hypothesis remains untested.
Ca2+-activated Cl− channels (CaCC) have also been implicated in the hypertensive remodeling process, and the identity of CaCC has also been difficult to define. Using 2K2C rats Wang et al. studied the involvement of the TMEM16 family of transmembrane proteins in regulating Ca2+-activated Cl− currents (ICl.Ca); in particular they focused on TMEM16A. The channel activity and ICl.Ca current were reduced in smooth muscle cells from hypertensive arteries compared to arteries from normotensive rats. Angiotensin II suppressed TMEM16A expression, and overexpressing TMEM16A impaired angiotensin II-mediated smooth muscle cell proliferation [123]. These studies suggest the TMEM family proteins may counterbalance artery remodeling involving smooth muscle cell hyperplasia in experimental models of hypertension. The TMEM proteins are possible therapeutic targets for vascular disease.
7.3 Inflammation
There is little doubt that low grade inflammation plays a significant role in the development of hypertension (for reviews see [124–126]) and many studies suggest that T cells are important mediators of the hypertension-associated vascular injury [126–128]. The effects of T cells on the cerebral arteries have not been well described. Macrophages have also been deemed to be important modulators of hypertension-associated vascular injury [129–131]. Recent studies from our laboratory show that macrophage infiltration is an important determinant of cerebral artery remodeling in SHRSP. In this study the peripheral macrophages were chronically depleted in young (6 week old) rats, the rats were studied at 12 weeks of age. Peripheral macrophage depletion caused a 50 % reduction in the number of perivascular macrophages surrounding the cerebral arteries. This caused a marked increase in middle cerebral artery lumen diameter and a reduction in the wall thickness and wall-to-lumen ratio. The cross-sectional area of the pial arteries was also increased by macrophage depletion. The change in middle cerebral artery structure had no effect on tone generation, or 5-HT induced contraction. However, it did cause an improvement in endothelium-dependent dilation that was associated with improved nitric oxide (NO) production. Importantly macrophage ablation did not lower blood pressure [132]. This study suggests that as hypertension develops, peripheral macrophages infiltrate into the cerebral vasculature and that these macrophages drive the hypertension-associated artery remodeling process.
Macrophages release multiple cytokines that could be involved in cerebral artery remodeling. One of the key candidates is tumor necrosis factor (TNF)-α which is involved in hypertension-associated renal injury [133, 134]. TNF-α inhibition with etanercept improved the middle cerebral artery structure in SHRSP. As with the study described above, rats were treated from 6 to 12 weeks so that the actions of TNF-α were being inhibited as the hypertension developed. TNF-α inhibition significantly increased the lumen diameter and reduced the wall thickness and wall-to-lumen ratio of the middle cerebral artery. Combined these changes in artery structure were associated with an improvement in pial artery perfusion [135].
8 Hemodynamic Effects of Artery Remodeling
Artery remodeling has several hemodynamic effects; in hypertensive subjects, artery remodeling is initiated in an effort to normalize cerebral blood flow, which increases as blood pressure increases. Artery remodeling normalizes flow by increasing vascular resistance [136, 137]. Therefore artery remodeling may be responsible for the relatively normal cerebral blood flow observed in hypertensive rats and patients. However, this situation changes as hypertensive subjects age; older hypertensive patients have reduced blood flow in specific brain regions including the occipitotemporal and prefrontal cortex, and the hippocampus [138]. Similarly, patients with poorly controlled blood pressure exhibit significant reductions in their total cerebral blood flow as they age. This cerebral hypoperfusion occurs independently of atherosclerosis [139], and could be the result of artery remodeling and impaired control of constriction and dilation. Aging is a major risk factor for cerebrovascular disorders independently of hypertension. But the incidence of hypertension in the elderly is approximately 70 % [140], which makes a clear separation between these two risk factors difficult. Clinical studies from the 1980s and early 1990s showed a positive correlation between white matter changes, a clinical marker of dementia, and thickening of the wall in small parenchymal arterioles [141, 142] suggesting the link between changes in cerebral artery structure and age-associated dementia. In aged Fischer F344 rats, there is inward hypotrophic remodeling and increased stiffness in pial arterioles [143]. In RORO rats, an inbred strain derived from Wistar rats with high longevity, the media thickness of the basilar artery is reduced in 32-month-old females, but not in 18-month-old females, without changes in lumen diameter [144]. In elderly humans, intracranial posterior arteries show inward eutrophic remodeling coupled to loss of compliance and elasticity [145]. A final structural alteration in aged intracranial arteries is the presence of various degrees of atherosclerosis, which can increase the risk of middle cerebral artery strokes [146].
9 Cerebral Artery Function in Hypertension
While the passive structure of the cerebral arteries contributes to the control of cerebral blood flow it is important to note that several other active mechanisms are involved in the regulation of cerebral perfusion. Studies of how cerebral arteries constrict and dilate are important. We know a lot about the mechanisms of constriction and dilation in cerebral arteries; we know much less about how hypertension affects these vasoactive mechanisms. Neurovascular coupling is the mechanism that links neuronal activity to regional cerebral perfusion. The cellular control of neurovascular coupling is complex and requires integrated signaling from neurons, interneurons, perivascular nerves, glia, and the cells within the vasculature [147, 148]. During neurovascular coupling vasoactive agents are released from active neurons, interneurons, astrocytes, and arteries themselves to produce localized vasodilation in parenchymal arterioles and upstream pial arteries; this in turn increases blood flow to the active neurons [24]. Human studies suggest that neurovascular coupling is impaired by hypertension [136].
Neurovascular coupling in rodents is commonly assessed by measuring the increase in blood flow or the increase in artery lumen diameter in response to various sensory stimuli including whisker stimulation. Studies using rodent models of hypertension suggest the hypertension-associated impairment in neurovascular coupling is more nuanced than just being a response to increased blood pressure. In fact increasing blood pressure in a mouse with phenylephrine does not affect neurovascular coupling suggesting that elevated blood pressure alone does not impair the increase in perfusion associated with neuronal activation [149]. Most studies suggest that activation of the renin angiotensin pathway is required to observe impaired neurovascular coupling in hypertensive animal models. Acute and chronic angiotensin II administration reduces the increase in cerebral blood flow observed with whisker simulation in mice [149]. However, this angiotensin II-mediated event is not blood pressure dependent because direct application of angiotensin II to the cerebral cortex has similar effects on cerebral blood flow in the absence of an increase in blood pressure [150]. The argument for a direct effect on angiotensin II on the brain is strengthened by studies showing that neurovascular coupling is impaired before the blood pressure becomes elevated mice given a low dose of angiotensin II that increases the blood pressure slowly [150]. The impaired neurovascular coupling observed in response to angiotensin II appears to require the activation of the type 1 angiotensin receptor and an increase in reactive oxygen species generation [151].
The effects of hypertension on neurovascular coupling in genetically hypertensive rats are less clear and less well studied. Studies using SHR show that the response to whisker simulation is impaired in rats with developed hypertension. Both the maximum increase in blood flow and the duration of the increase in blood flow were reduced in the SHR. Surprisingly, treating the rats with losartan to lower the blood pressure did not improve the functional impairment in neurovascular coupling and the impaired blood flow was not associated with reductions in vessel density or artery lumen diameters [152]. This suggests that in rats with sustained polygenic hypertension angiotensin II may not be the sole determinant of the impaired neurovascular coupling. The reason why blood pressure lowering does not improve the blood flow response to simulation is less clear and may be associated with long-term dysfunction of the cerebral arteries.
10 Cerebral Artery Autoregulation
The cerebral circulation is endowed with the innate ability to keep parenchymal perfusion constant while the perfusion pressure is fluctuating; this occurs through the mechanism of autoregulation, also known as Bayliss effect (Fig. 6.2). The autoregulatory range is the range of blood pressure over which the cerebral blood flow remains constant. In most adults this range is between mean arterial pressures of 50–60 mmHg and 150 mmHg [153]. Blood flow control is lost when the perfusion pressure is outside the autoregulatory range; in this situation the cerebral blood flow is dependent on the mean arterial pressure [154]. Pressures above the autoregulatory range cause increased cerebral perfusion and eventually vasogenic edema; pressures below the autoregulatory range cause cerebral hypoperfusion and ischemic injury [11]. Studies to investigate the factors that control autoregulation have been fruitful. Myogenic reactivity is an important determinant of cerebral autoregulation (this will be discussed further below). Neuronal NO production modulates myogenic reactivity and therefore autoregulation [153, 155–157]. Recent studies in humans suggest that sympathetic vasoconstriction [158] and cholinergic vasodilation [159] play an important role in autoregulation. Autoregulation is also controlled by blood flow itself; Koller and Toth recently reviewed this concept [160], and there is evidence suggesting that increased flow causes constriction or dilation depending on the artery being studied.
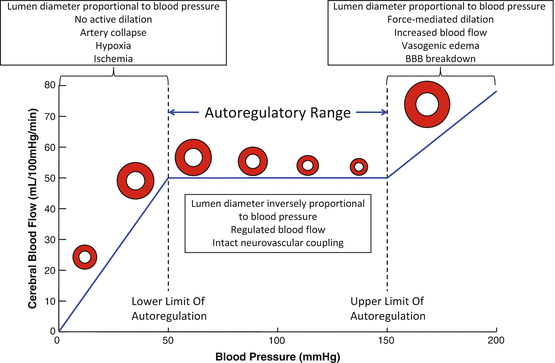
Fig. 6.2
Cerebral blood flow and autoregulation. The lower and upper limits of cerebral blood flow autoregulation are indicated by the dotted vertical lines. The blue line depicts cerebral blood flow, the red circles above this line provide an indication of the effects on the increased intraluminal pressure on the lumen diameter of the cerebral arteries
11 Myogenic Reactivity
Before discussing myogenic reactivity it is necessary that we define some key terms. Myogenic reactivity is the ability of the artery to change its tone (or degree of constriction) to respond to fluctuations in intraluminal pressure while keeping blood flow constant [161]. Myogenic tone is an intrinsic property of arteries and arterioles to maintain a spontaneous active contractile force in the smooth muscle cells. Several factors contribute to myogenic tone regulation; these include the intraluminal pressure, resting potassium conductance, calcium channel activity, and the sensitivity of the smooth muscle cell contractile machinery to calcium. Tone is also regulated by extrinsic factors, such as hypoxia.
As mentioned previously, myogenic reactivity is key determinant of cerebral autoregulation; this is particularly true at higher perfusion pressures when arteries constrict to maintain blood flow at a constant level [154]. Cerebrovascular resistance is regulated by basal vascular tone, and much of this tone is myogenic [162]. Myogenic tone is increased when the endothelium is removed from arteries [163, 164] and the myogenic response and myogenic reactivity are regulated by the endothelium through several mechanisms. Myogenic constriction is reduced by agents produced in the endothelium, including NO [165], prostacyclin [166], and endothelium derived hyperpolarizing factor [167]. Conversely, 20-HETE may increase myogenic tone and its production is increased in arteries from hypertensive rats [168, 169].
Pressure myography is commonly used to study the myogenic response. Using this technique the responses of the artery to pressure and stretch can be studied in the absence of confounding factors such as circulating vasoactive compounds and metabolites. The myogenic behavior of large cerebral arteries occurs in three stages. Tone develops in phase one; this requires smooth muscle cell depolarization and occurs at intraluminal pressures of 40–60 mmHg. Intracellular calcium is increased during this stage, and tone development reduces artery wall tension and tangential stress. Phase 2 is myogenic reactivity and this occurs between the intraluminal pressures of approximately 60 and 140 mmHg. In this phase the tone that developed in phase 1 is maintained even though the wall tension and intraluminal pressure are increased. Phase 3 is force-mediated dilation, which occurs when the intraluminal pressure increases to the point where the tangential wall stress exceeds the smooth muscle cells ability to contract [154].
A few studies have assessed autoregulation and myogenic reactivity in models of hypertension, and they are conflicting. Pressure myograph studies show that posterior cerebral arteries from normotensive WKY rats exhibit myogenic reactivity over a range of intraluminal pressures from 40 to 150 mmHg with force-mediated dilation occurring at higher pressures. In SHR the range of pressures where myogenic reactivity occurs is higher (65 to 190 mmHg) [170], and the autoregulatory curve is shifted to the right. However, other studies show that myogenic tone generation is increased in posterior cerebral arteries from SHR compared to WKY rats; i.e., at each intraluminal pressure studied the arteries from the SHR were relatively more constricted than those from WKY rats. This increased tone generation occurred over a wide range of intraluminal pressures, but arteries from both strains experienced force-mediated dilation at the same pressure [171]. Middle cerebral arteries from male SHRSP show similar myogenic responses to WKY rats, but the arteries from female rats behave differently. Middle cerebral arteries from female SHRSPs exhibited more myogenic constriction than female WKY rats [172]. Recent studies from Chan et al. show a small but insignificant increase in tone generation in the penetrating arterioles from female SHR compared to WKY rats [74]. These studies were conducted in 18-week-old rats, and it is possible that statistical significance would be reached in older rats.
Feeding SHRSP a high-salt diet causes the middle cerebral arteries to lose their ability to generate myogenic tone [173]. In this situation the downstream arterioles and capillaries are at risk of rupture because of the elevated blood pressure. SHRSP fed a high-salt diet from weaning begin developing hemorrhagic strokes at about 13 weeks of age—the ability of the cerebral arteries to autoregulate is also completely lost at this time, and a pressure-dependent increase in cerebral perfusion is observed [174]. The mechanisms responsible for the loss of myogenic capacity in salt-loaded SHRSP have not been elucidated. It is possible that this is a physical response to the exacerbation of hypertension that occurs with the administration of a high-salt diet to SHRSP [175, 176].
Aging alters the effects of hypertension on cerebral autoregulation. Young (3-month-old) or aged (24-month-old) mice were treated with angiotensin II to develop hypertension, and the myogenic reactivity of the middle cerebral artery was studied. The middle cerebral artery from young angiotensin II hypertensive mice showed the classical increase in tone associated with elevated intraluminal pressures. Myogenic reactivity was lost in the middle cerebral arteries from hypertensive, aged mice. The loss of myogenic reactivity was attributed to impaired 20-HETE production and reduced activity of the TRP channel canonical 6 (TRPC6), which are responsible for maintenance of myogenic responses in cerebral arteries [177]. 20-HETE and TRPC6 contribute to an increase in intracellular Ca2+ in smooth muscle cells, leading to constriction in middle cerebral arteries from young, but not from aged, angiotensin II hypertensive mice [178].
The ability of an artery to respond to increases in intraluminal pressures requires that the arteries express some kind of mechanosensor that will link pressure and stretch to constriction. The precise identity of the mechanosensors in the cerebral vasculature is the subject of much debate, but a few candidates have emerged in the recent years. TRPC6 mediate myogenic constriction of cerebral arteries [179]. Similarly, TRP melastatin (TRPM) 4 regulates myogenic tone and reactivity in large intracranial arteries [180]. The currently accepted model of mechanotransduction proposes that membrane stretch causes angiotensin II receptor type 1 activation and Src-tyrosine kinase-dependent activation of PLCγ1. PLCγ1 activation generates IP3, which activates IP3 receptors in the sarcoplasmic reticulum, leading to release of Ca2+ that activates TRPM4 in the membrane, resulting in Na+ influx, depolarization, and smooth muscle cell constriction. At the same time, membrane stretch activates TRPC6, resulting in Ca2+ influx that positively modulates IP3 receptor activity [181]. TRPM4 are also important regulators of myogenic activity in parenchymal arterioles, although it seems in these arterioles that the mechanosensors are P2Y receptors [182]. The effect of hypertension on these mechanosensor pathways has not been studied. It is possible that the activity of these pathways is increased in arteries from hypertensive subjects, and this could account for the increase in myogenic tone and reactivity.
While the upper end of the autoregulatory curve has received a lot of attention we know much less about how hypertension affects the lower end of the curve and we may be missing important physiological effects. The lower limit of autoregulation is the pressure below which blood flow becomes dependent on blood pressure. When the intraluminal pressure falls below this level active dilation no longer occurs and the arteries collapse because the intraluminal pressure is low; this causes blood flow to fall. In animals with hypertension the lower limit of autoregulation is increased [183] and lowering blood pressure with an ACE inhibitor reduces the lower limit of cerebral blood flow regulation [87, 184, 185]. Reducing the lower limit of blood flow regulation may be important in situations like cerebral ischemia. When a major artery is occluded the intraluminal pressure in arteries downstream from the blockage drops—this would normally cause vasodilation, but if the lower limit of autoregulation is breached, the diameter of the artery, and therefore blood flow, will be dependent on its passive diameter. In this situation the inward remodeling observed in cerebral arteries from hypertensive rats becomes particularly detrimental because it limits perfusion.
12 Regulation of Cerebral Artery Tone: Nitric Oxide-Dependent Mechanisms
NO generation is the mechanism that has been best described for the regulation of cerebral artery tone (Fig. 6.3). Endothelial NO synthase (eNOS) is an important source of NO in cerebral arteries; cerebral microvessels from SHR have lower eNOS expression than those from WKY rats [83], and the impaired NO production associated with this reduces endothelium-dependent dilation, which could lead to increased hypoxia and neuronal death after ischemia. Under normal conditions the arteries around an ischemic occlusion dilate to improve blood flow to the region and reduce the ischemic injury. In hypertensive rats this protective mechanism is impaired [186] and this may be the result of impaired NO production. This hypothesis was tested in studies using eNOS knockout mice. These mice have larger cerebral infarcts after the induction of ischemia than control mice and this appears to be the result of impaired cerebral perfusion [187].
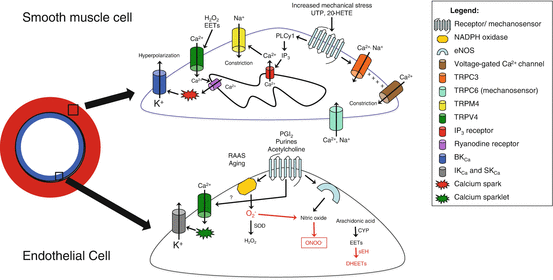
Fig. 6.3
Novel signaling mechanisms controlling cerebral artery function. The endothelial cell is shown below the vascular smooth muscle cell with a diagram of the location of these cells in the artery on the left of the figure. The following major recent findings are summarized: (1) P2Y receptors and TRPC6 channels are possible mechanosensors, leading to increases in IP3, generation of a calcium spark, and opening of TRPM4 channels, causing depolarization and activation of voltage-gated Ca2+ channels [179–182]; (2) TRPC3 channels, found in smooth muscle cells, are overexpressed in hypertensive carotid arteries and are linked to UTP-evoked constriction. Opening of TRPC3 channels causes a localized depolarization that opens L-type Ca2+ channels, leading to depolarization and vasoconstriction [204, 205]. (3) TRPV4 channels in endothelial cells are downstream to muscarinic receptor activation and generate a calcium sparklet, which opens IKCa and SKCa channels, leading to K+ efflux and hyperpolarization [210, 211]; (4) EETs activate TRPV4 channels, causing a small calcium influx that opens RyRs in the sarcoplasmic reticulum. RyR are permeable to Ca2+, generating a calcium spark, which is coupled to opening BKCa, leading to efflux of K+ and hyperpolarization [206, 207]. The soluble epoxide hydrolase (sEH) inactivates EETs to dihydroxyeicosatrienoic acids (DHETs) and may be increased in hypertension (red) [208, 209]; (5) many vasoactive peptides, including members of the RAAS, increase the activity of the enzyme NADPH oxidase, causing accumulation of superoxide. Superoxide can be dismutated into hydrogen peroxide by the superoxide dismutase enzyme, or it can react with NO, blunting NO availability. Cyp cytochrome P-450, RAAS renin-angiotensin-aldosterone system
Some drugs have the potential to increase eNOS activity and these drugs also reduce the damage caused by ischemia. Cilostazol is a phosphodiesterase-3 inhibitor used to treat peripheral vascular disease and intermittent claudication. Cilostazol increases eNOS phosphorylation, a marker of enzyme activation, in brains from SHR; this increase in eNOS activation was associated with increased cerebral perfusion after the induction of ischemia [188]. In SHRSP fed a high-fat diet, cilostazol increased cerebral blood flow compared to untreated SHRSP and SHRSP treated with aspirin or clopidogrel. The absence of an effect of aspirin or clopidogrel suggests that the beneficial effects of cilostazol on cerebral blood flow are not merely a function of its antiplatelet activity [189], and that the enhanced perfusion may be the result of improved endothelial function. Clinically this finding could be important. The Cilostazol Stroke Prevention Study showed that cilostazol treatment prevents secondary ischemic strokes; interestingly, this effect was found to be independent of cilostazol’s effects on platelet aggregation [190] and may therefore be related to improved cerebrovascular health.
Increased oxidative stress causes impaired NO-dependent dilation in arteries from hypertensive subjects [191]. Under normal physiological conditions, superoxide dismutase converts superoxide to hydrogen peroxide. Excessive superoxide production saturates the cell’s defenses, which increases the superoxide available to react with NO and reduces its bioavailability [192]. This pathway may be particularly important in cerebral arteries because they have an enhanced capacity to produce superoxide when compared to peripheral arteries [193]. This may be because reactive oxygen species play a physiological role in the cerebral circulation. For example, hydrogen peroxide causes pial artery dilation by activating large conductance Ca2+-activated potassium channels (BKCa) [194]. Conversely, in the middle cerebral artery, hydrogen peroxide causes constriction by opening L-type voltage-gated Ca2+ channels [195]. Interestingly, there is little superoxide production in basilar arteries from SHR. This may be the result of enhanced superoxide dismutase expression since superoxide was detected after inhibition of superoxide dismutase [196]. The mechanism responsible for the increased superoxide dismutase expression is unknown, but it may be a response to enhanced superoxide production, which may function as an alternative vasodilator mechanism during chronic NO depletion (Fig. 6.3).
13 Regulation of Cerebral Artery Tone: Endothelium-Dependent, Nitric Oxide-Independent Dilation
Endothelial-dependent hyperpolarizing factor (EDHF) (Fig. 6.3) is responsible for NO and prostacyclin-independent vasodilation [197]. The identity of EDHF is the subject of intense debate, but calcium-activated K+ channels, the small- and intermediate-conductance Ca2+-activated K+ channels (SKCa and IKCa, respectively) in particular, are involved in EDHF-mediated dilation [198]. IKCa knockout mice develop hypertension and acetylcholine-mediated dilation is impaired in carotid arteries from these mice [199]. In middle cerebral arteries EDHF-dependent dilation requires IKCa activity [200]. It is unclear if IKCa expression is reduced in cerebral arteries from hypertensive subjects. Interestingly, IKCa expression is elevated in mesenteric resistance arteries from SHRSP, but EDHF-mediated dilation is impaired [201]. This study suggests that the IKCa channel is not solely responsible for EDHF-mediated dilation.
TRP channels are increasingly being recognized as important regulators of cerebral artery endothelial function (Fig. 6.3). TRP channels are nonselective for cations, but some channel subtypes are partially selective for monovalent or divalent cations (for a review see [202, 203]). The TRPC3 is expressed in cerebral arteries and TRPC3 activation causes vasoconstriction [204]. TRPC3 expression is increased in carotid arteries from SHR and this is linked to augmented contractility [205]. Similarly, increased TRPC3 expression and activity was shown in mesenteric arteries of SHR and linked to higher contractility to endothelin-I. Although TRPC3 expression in cerebral arteries from hypertensive rats has not been studied, it is possible that there is an increase in TRPC3 activity to enhance myogenic constriction.
TRPV4 channels are activated by epoxyeicosatrienoic acids (EETs) and this causes endothelium-dependent dilation in cerebral arteries [206, 207]. The soluble epoxide hydrolase inactivates EETs to dihydroxyeicosatrienoic acids and may be increased in hypertension [208, 209]. TRPV4 activation causes smooth muscle and endothelial cell hyperpolarization, but the mechanism responsible for this in the two cell types differs. In vascular smooth muscle cells, TRPV4 channels form a complex with ryanodine receptors and large conductance Ca2+-activated K+ (BKCa) channels [206]. TRPV4 channel activation allows for a small Ca2+ influx; this leads to ryanodine receptor activation in the sarcoplasmic reticulum and the generation of calcium sparks, which are transient and localized increases in Ca2+. The calcium sparks cause an increase in BKCa opening and this leads to K+ efflux and hyperpolarization [206]. In mesenteric resistance artery endothelial cells, muscarinic receptor activation increases TRPV4 channel opening; this generates calcium sparklets, which are transient increases in intracellular calcium in close proximity to a single channel [210]. Calcium sparklets activate IKCa and SKCa channels and this causes endothelial cell hyperpolarization that is transmitted through gap junctions to the smooth muscle [211]. Calcium sparklets also occur in smooth muscle cells but they are not linked to TRPV4 activity. Reactive oxygen species activate Ca2+ sparks in cerebral arteries, coupling this signal to ryanodine receptors and BKCa [212]; this may be the result of TRPV4 activation. TRPV4 channels play an important role in regulating endothelial function in cerebral arteries; however, the expression and function of the TRPV4 channel has not been studied in cerebral arteries from hypertensive subjects. TRPV4 channel knockout mice do not develop spontaneous hypertension, but when the mice are treated with l-NAME the resultant hypertension is exacerbated. This enhanced blood pressure response has been linked to impaired endothelium-dependent dilation in peripheral arteries [213]. Recent studies have implicated reactive oxygen species generation by NADPH oxidase in the impaired basilar artery endothelial function in DOCA-salt hypertensive mice [92].
Related posts:
 Hypertension and Brain Inflammation: Role of RAS-Induced Glial Activation
Hypertension and Brain Inflammation: Role of RAS-Induced Glial Activation
 A Systematic Review of Reviews to Examine the Association Between Hypertension and Cognitive Impairments in Adults
A Systematic Review of Reviews to Examine the Association Between Hypertension and Cognitive Impairments in Adults
 Arterial Stiffness and the Brain
Arterial Stiffness and the Brain
 Endothelium, the Blood–Brain Barrier, and Hypertension
Endothelium, the Blood–Brain Barrier, and Hypertension
 Cognitive Dysfunction and Dementia in Animal Models of Hypertension
Cognitive Dysfunction and Dementia in Animal Models of Hypertension
 Hypertension and Stroke
Hypertension and Stroke
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


