Calculation of verbal fluency index. The verbal fluency index (Vfi) is calculated using a motor control condition in which after the participant has generated words in the given time (e.g., one minute to generate words beginning with a given letter), they are then asked to copy out or read aloud the words they have previously generated as fast as possible. This gives a measure of motor speed. The total time for the test minus the measure of motor speed is then divided by the number of words produced to give an estimation of the average time taken to “think” of each word or Vfi.
Executive dysfunction has been revealed on a range of tests including tests of attention monitoring and switching, rule deduction, cognitive flexibility, such as the Wisconsin Card Sorting or Trail Making Tests [35, 36, 39], and on other measures of concept formation (Delis–Kaplan Executive Function System Sorting Test), with poor performance in some ALS patients which was related to reduced verbal fluency [40]. Furthermore, deficits have also been revealed on tests dependent on the manipulations of concepts in working memory, such as reverse digit span or the N-back task. Impairments have also recently been shown using dual-task paradigms [41]. This procedure is thought to tap functions of the central executive component of working memory by performing two tasks concurrently (in this case a visual processing speed task and digit recall). Of note patients did not show slowed processing speed using a visual inspection time task in which stimulus presentation times were altered and which was therefore independent of motor speed. Thus, this study demonstrated executive dysfunction without generalized cognitive slowing.
The neuropsychological literature demonstrates that most of these executive functions are mediated by dorsolateral prefrontal cortex (see imaging section for further support). However other functions that are more dependent on orbitomedial prefrontal processes are now the focus of investigation. Patients have been shown to have abnormal risk-taking behavior on the Iowa Gambling Task with a failure to learn to avoid high-risk choices of decks of cards on the basis of monetary rewards or punishments [42]. Other studies have employed tasks which purportedly are more ecologically valid than traditional tests of executive functions in an attempt to relate dysfunction to everyday life. Deficits in ALS patients have been shown using the Medication Scheduling Task [43] and the Holiday Apartment Task [44] with patients showing difficulties in reasoning and coordinating rules. The latter consists of a non-risk decision-making task which involves mental heuristics. ALS patients’ strategy use on this task appeared akin to that of patients with damage of the ventromedial prefrontal cortex and not those with dorsolateral prefrontal dysfunction.
Language dysfunction in FTD-ALS
Although language symptoms were frequently discussed in early description of ALS [1], they were generally considered, until the seminal paper by Caselli et al. [45], to play only a marginal role in the disease. One of the biggest problems impeding the study of language in ALS is the fact that a large proportion of patients present with dysarthria or dysphonia as a consequence of the involvement of speech-controlling muscles. Hence, the dramatic reduction in speech output seen in many ALS patients tended to be attributed to a peripheral impairment rather than to a central involvement of the faculty of language. Furthermore, formal language assessment was rarely conducted.
Advances in our understanding of the language impairments in ALS came from two directions. One was the study of language comprehension. Tests using tasks such as pointing to a picture allowed researchers to examine the understanding of single words as well as full sentences even in patients with practically absent spontaneous speech output. The emerging evidence from this work highlighted not only pronounced difficulty in understanding grammatically complex sentences but also a less expected deficit in the comprehension of single words, particularly verbs [46]. Since then, deficits in the processing of verbs and also the underlying concepts of actions have become one of the most consistently documented linguistic features of ALS [47–49] and have been associated with characteristic pathologic changes in Brodmann areas 44 and 45 (Broca’s area) [4]. Indeed, ALS has come to be regarded as a prototypical “lesion example” illustrating theories of embodied cognition [50, 51].
The second area which proved particularly fruitful in ALS research is the analysis of written language. Like the tests of comprehension based on pointing rather than speaking, written language can be studied also in patients with severe dysarthria or even mutism. As in language comprehension, the examination of written language has unearthed impairment at the single-word as well as sentence level [4]. A pioneering work in this field was conducted by Japanese authors who demonstrated that ALS patients tend to have more problems with the phonologic kana than with the semantically related kanji script [52]. This is remarkable since kana is visually much simpler, has fewer characters, and is acquired earlier developmentally than kanji. One would expect, therefore, that kana would be more resistant to brain pathology than the more complex kanji. The reason for this has not yet been determined, but interestingly spelling errors have now also been documented in English-speaking patients [53].
Considering these two areas of language comprehension and written language, it is not surprising that language impairment has been so long overlooked in ALS. The most common test of language functions in clinical practice, picture naming, is of limited value in ALS patients. First, it relies on speech production, which, as was mentioned before, is often reduced or even absent. Second, the vast majority of naming tests use only pictures of objects, and hence are not able to detect deficit in verb processing characteristic for ALS. Accordingly, a recent study using a much wider range of language tasks than most previous ones estimated that language dysfunction might constitute the most common cognitive deficit in ALS patients, even more common than executive dysfunction [18, 54]. In practical terms, comprehension of verbs as well as spelling are now part of a new screening test for cognitive deficits in ALS (the Edinburgh Cognitive and Behavioural ALS Screen), which will be described in more detail later.
When we compare the profile of language dysfunction described in this chapter with that of the two aphasic variants of FTD (PNFA and SD), it becomes clear that they demonstrate only partial overlap. The mute or almost mute ALS patients are different from those with PNFA, who often struggle for words but make every possible effort to produce them. The prominent impairment of verb and action processing in ALS shows the opposite pattern of predominant noun and object deficit in SD [55], although PNFA patients may show verb-predominant deficits. Thus, we believe that the language characteristics of FTD-ALS are distinguishable from those of the aphasic subtypes of FTD [1, 50], but further comparative investigation is warranted.
Social cognition in FTD-ALS
In the last five years neuropsychologists have directed their attention toward the investigation of changes in social cognition in ALS, given that this is a primary feature of FTD, with a particular focus on theory of mind and emotional processing. Theory of mind tests involve the ability to infer mental states in others. ALS patients have been shown to be impaired in the interpretation of social scenarios in stories and humorous cartoons, and in the understanding of faux pas through written stories [44]. A selective deficit in processing specifically social cartoon scenes was demonstrated by Cavallo et al. [56], in which ALS patients had particular difficulties in inferring a social intention compared with a private or “non-social” intention. A private intention involves a goal that is only relevant to one person (e.g., the person wants to read a book), compared with a social goal (e.g., the person wants another person to move their bag so they can sit down). A deficit has also been revealed on a simple theory of mind test in which the participant must infer the thoughts of another (depicted by a cartoon face) by the direction of eye gaze [42]. Here the participant must first choose their own favorite object from a choice of four objects; a central face then appears on the screen which is looking and smiling at one of the objects and the participant must choose which object the face likes best. ALS patients tend to repeatedly choose their own favorite object, indicating a difficulty in inhibiting their own preference, rather than the normative response of using a simple social cue of eye gaze to understand the perspective of another person.
ALS patients also show difficulties in processing emotions. Impairments have been revealed in the recognition of facial emotional expressions, and judgments of approachability on the basis of facial expressions [42, 57]. Furthermore, deficits have been found using the Reading the Mind in the Eyes Test and also in judgments of emotional prosody [44]. Lulé et al. demonstrated that ALS patients tend to show a more positive valence towards emotive social situations and overall a more balanced state of arousal than controls, rating calm pictures as more exciting and vice versa [58], while Papps et al. revealed that ALS patients do not show the enhanced recognition of emotional words as found in healthy controls [59].
Behavior change in FTD-ALS
In their influential paper, Lomen-Hoerth et al. demonstrated that new-onset behavior change similar to that found in bvFTD was prevalent in ALS [60]. As measured using the Neuropsychiatric Inventory, apathy, disinhibition, and poor social monitoring were reported both in cases with letter fluency deficits and in others who had no evidence of cognitive change. In a detailed interview-based assessment of behavior change, self-centeredness/selfishness was described as the most prominent symptom followed by apathy, aggression, loss of insight, and social disinhibition. Apathy appears to be a particularly common behavioral feature of ALS and has typically been measured using the Frontal Systems Behavior Scale (FrSBe) [42, 61], although this assessment method has limitations and may exaggerate impairment due to physical disability. However, evidence for prevalent apathy in ALS has also been shown using the Cambridge Behaviour Inventory in which this symptom was reported in 41% of patients [62]. An ALS-specific behavior questionnaire has been designed which detected a lower prevalence of behavior change than the FrSBe although behavior change was still evident [63].
Psychiatric symptoms in FTD-ALS
As discussed earlier in this chapter, much of our current understanding of the FTD-ALS spectrum brings us back to the historical insights of the neurologists and psychiatrists of the early twentieth century. This is particularly true for the psychiatric features of the disease. Many symptoms mentioned in the classical papers on ALS, such as personality change, irritability, or emotional lability, could now be reinterpreted as belonging to the spectrum of bvFTD. However, several early descriptions also explicitly mention psychotic symptoms, specifically hallucinations and delusions, and speak of schizophrenia associated with ALS (for review see [1, 15]). More recent papers, focusing on other aspects of the disease, such as language, mention complex delusions, such as the “phantom lodger syndrome” [4]. Such psychotic symptoms are relatively rare in the classical FTD [64], so they constitute yet another difference between FTD and FTD-ALS. Indeed, the appearance of delusions in FTD patients is associated with a significantly higher risk of subsequent development of ALS [5].
The question of psychotic symptoms in FTD-ALS acquired a new dimension with the finding that delusions and hallucinations were particularly frequent among the patients with the C9orf72 mutation [3]. But why have only some of the research groups found such an association and others not? This question was intensely debated at the Biennial Meeting of the World Federation of Neurology Research Group on Aphasia, Dementia and Cognitive Disorders (WFN RGADCD) in Hyderabad in December 2012. One option is that the patient cohorts examined at different research centers are indeed different. The other, suggested by one author of this chapter (THB), is that the difference lies in the assessment of patients. As mentioned before, most patients with FTD-ALS show a characteristic temporal pattern of the disease, starting with psychiatric symptoms and then followed, months or years later, by cognitive and finally amyotrophic symptoms. So in many if not in most cases the psychotic symptoms are not present any more at the time the diagnosis of ALS is made. Patients and families would not likely recognize a connection between two features which appear as dissimilar as paranoia and muscle weakness, so the history of psychotic symptoms goes unnoticed. The only way to determine real prevalence of psychosis is a systematic enquiry. For this reason, our new screening instrument, which will be presented in detail below, contains, apart from the questions exploring the classical symptoms of bvFTD, also specific inquiry about a history of psychotic symptoms.
Assessing cognitive and behavior dysfunction in FTD-ALS
Despite the increased awareness of cognitive change as integral to the disease [24], the cognitive status of the majority of ALS patients attending clinics remains unknown [2, 54]. This reflects the orientation of many ALS neurologists (not recognizing the frequency of these symptoms) and the lack of expert clinical neuropsychology services associated with ALS or neuromuscular clinics; in addition, there has been a dearth of suitable screening tools for clinical use. Furthermore, the diversity of physical disability in ALS necessitates the use of specifically developed measures as well as expertise. Standard tests may not be suitable and may exaggerate performance deficits due to physical limitations.
Two cognitive screening examinations have been developed specifically for ALS. The first, the ALS Cognitive Behavior Screen (ALS-CBS) [65] is a very brief screen which assesses a single cognitive domain of executive functions. It contains eight short cognitive tests of executive functions and a carer behavior questionnaire. It has been validated against a neuropsychological battery and successfully distinguishes those with cognitive impairment from those with no cognitive impairment with 85% sensitivity and 71% specificity. The second is the Edinburgh Cognitive and Behavioural ALS Screen (ECAS), which is a multidomain, brief (15–20 minute) assessment for use within an MND clinic setting, for use also by non-neuropsychologists, such as physicians, speech pathologists, nurses, or other clinicians [53] (see Figure 6.2). It was designed to determine: (1) Which patients have cognitive and/or behavior impairment? (2) How severe is that impairment? And most importantly given the heterogeneity of presentation (3) What type of cognitive or behavioral impairment is present? The ECAS assesses functions which are specific to the cognitive profile of ALS (including executive and language functions, social cognition, and behavior). It also includes brief assessment of functions not specific to the cognitive profile of ALS (memory and visuospatial functions) but which are typically affected in other disorders common in older adults, namely Alzheimer’s disease. Our recent work has demonstrated that of a cohort of 48 ALS patients (none with evident dementia), 29% were below cutoffs for abnormality on the ECAS total scores, with 35% showing abnormal language functions, and 23% executive and fluency deficits. The ECAS also includes a separate, brief carer behavior interview based on the recent criteria for diagnosis of bvFTD [9]. Forty percent of carers interviewed reported change in at least one behavioral domain, the most prevalent being apathy, which is in accord with previous findings. The cognitive and behavioral data from the ECAS are therefore consistent with prevalence rates demonstrated by studies using extensive neuropsychological batteries.
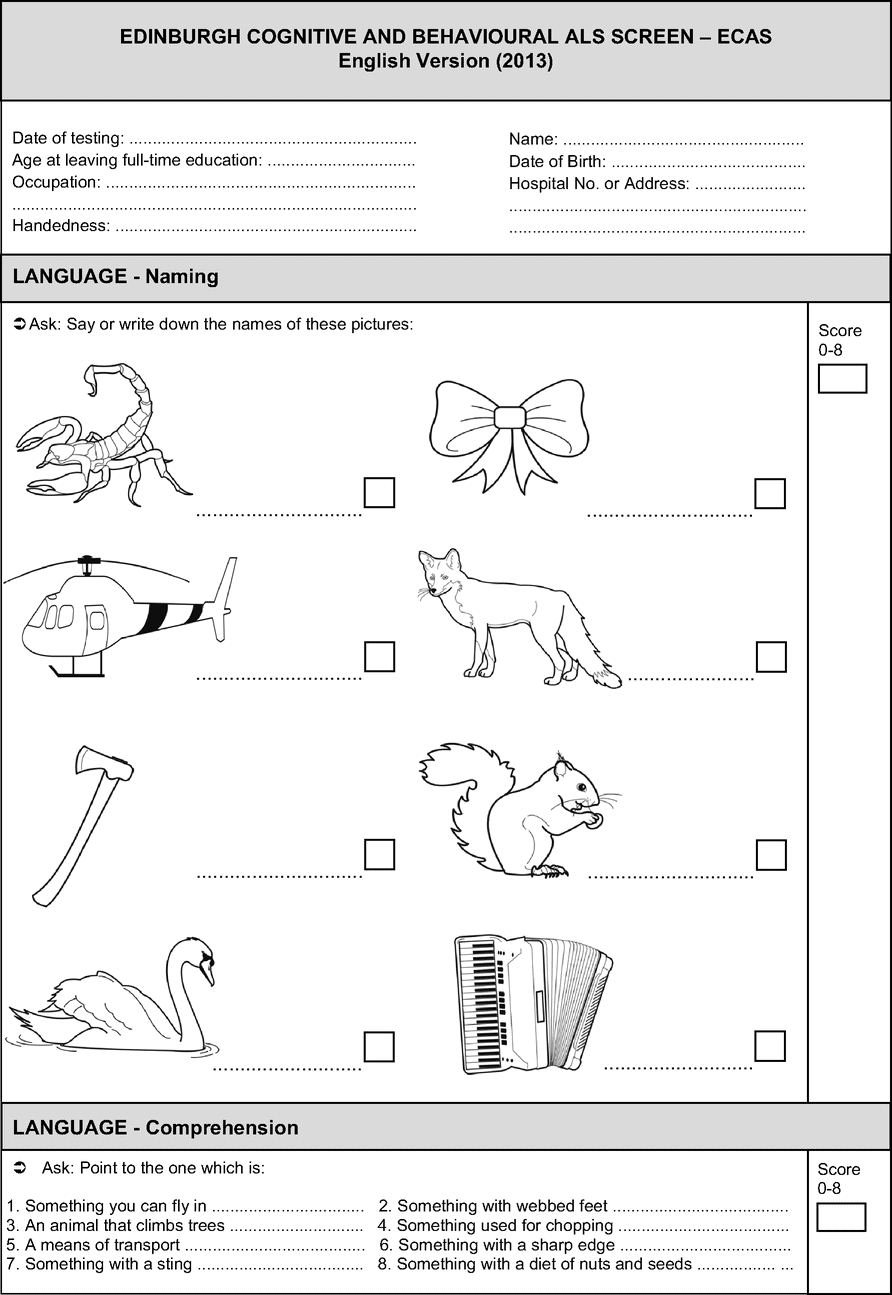
Why is it important to identify cognitive and/or behavioral symptoms in ALS? Cognitive change may impact on planning, attention, decision-making, and initiating ideas. Especially in the context of a condition as serious as ALS, clinicians must ensure that patients can fully understand the consequence of their decisions. Changes in behavior, personality, or social cognition may result in an egocentric perspective. After cognitive and/or behavior change is identified, carers, clinicians, and patients should be educated that these changes are part of the spectrum of symptoms of the disease. Hidden impairments may emerge as the patient faces new challenges in coping with their disability such as learning to use communication, feeding, or respiration aids. Difficulties with managing affairs or finances or end-of-life decisions may come to light. Furthermore, neurobehavioral symptoms have been associated with poor quality of life, increased depression and higher carer burden [66], while Lillo et al. [16] demonstrated that the strongest predictor of high caregiver burden was abnormal behaviors (such as disinhibition and impulsivity) which were over and above physical disability in ALS.
Brain imaging and cognition in FTD-ALS
A variety of imaging techniques have shown that cognitive symptoms in ALS are directly related to frontotemporal cortical and subcortical involvement [67–69]. Functional MRI (fMRI) and fluorodeoxyglucose positron emission tomography (FDG-PET) have shown that verbal fluency deficits are associated with dysfunction of the dorsolateral prefrontal cortex and anterior cingulate gyrus [37, 67, 70]. Impairments in attention and inhibition have also recently been investigated using fMRI, with a profile of abnormal activation in in the medial prefrontal cortex, anterior cingulate gyrus, and temporal cortex [71]. Correlations between impairments in verbal fluency and naming and reduced flumazenil PET binding in the inferior frontal gyrus have also been reported, indicating a focal reduction in GABA receptors [72].
Studies on structural abnormalities have recently focused on white matter changes. One of the earlier studies using automated volumetric MRI analysis with patients with verbal fluency deficits showed a pattern of frontotemporal white matter change [31]. Further correlations with white matter tract abnormalities have been revealed using diffusion tensor MRI in which verbal fluency impairments have been correlated with a reduction in fractional anisotropy in the cingulum bundle [73] and inferior frontal gyrus white matter and corpus callosum, the latter being a key feature consistently across studies [41] (see Figure 6.3).
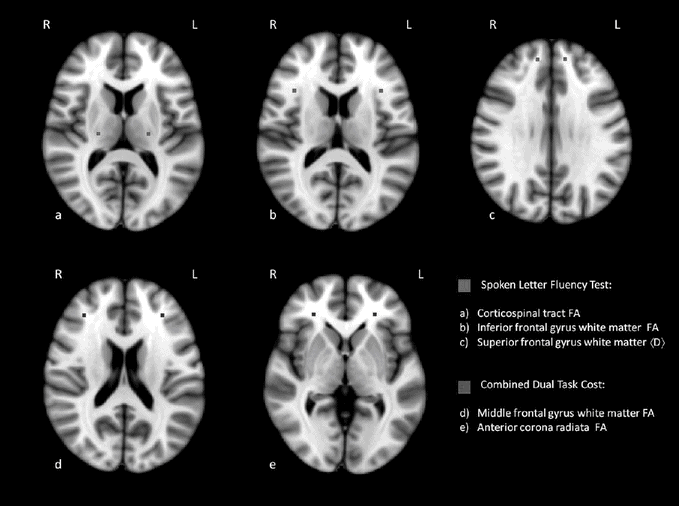
Cognitive performance correlates with white matter tract abnormalities using diffusion tensor MRI. Correlations between verbal fluency index (Vfi) and dual-task performance with fractional anisotropy (FA) and diffusivity, <D>, diffusion tensor imaging measures in prefrontal and motor system tracts in ALS [41].
Dual-task impairments appear more strongly correlated with dorsolateral prefrontal dysfunction while letter fluency is more dependent on inferolateral prefrontal dysfunction [41]. Impairments in emotional processing have also been investigated with brain imaging and levels of arousal have been shown to correlate with activation in the anterior insula in ALS [58], while right hemispheric dysfunction was implicated in ALS during an emotional decision-making and recognition task [74]. Furthermore, impaired emotional empathy has been related to reduced gray matter density in the anterior cingulate cortex and right inferior frontal gyrus [75]. Greater apathy has been related to dysfunction of the anterior cingulum in ALS [76], while cortical atrophy was shown to be linked to both neuropsychiatric and cognitive changes in ALS [77].
Pathology of FTD-ALS
At the level of pathology, FTD and ALS show striking similarities. In the 1990s it was recognized that the brains of patients with ALS as well as those of a large number of tau-protein-negative cases of FTD (including most cases of SD and a large proportion of those with bvFTD) showed similar intracellular ubiquitin-positive inclusions. In 2006, the protein TDP-43 was identified in such inclusions [12]. Another relevant protein, FUS (fused in sarcoma protein), was described three years later [78]. From the molecular point of view, therefore, it is compelling to view FTD and ALS as different manifestations of the same underlying processes. Their phenotypical variation can be explained by the different distribution of pathology (see Chapter 13 for more detail).
Like most other neurodegenerative diseases, FTD and ALS begin usually with a relatively focal presentation, followed by a gradual but relentless progression. The nature of this progression is a topic of considerable interest. Within the classical motor presentation of ALS, large systematic clinicopathologic studies by Ravits and colleagues showed a focal initiation and a spread of the disease to functionally connected structures [79]. A similar method of investigation was recently undertaken in relation to the spread of TDP-43 in ALS [80] and in bvFTD [81]. A possible explanation for the functional link between the cognitive and the motor system as an axis of spread of pathology has been offered by Bak and Chandran [50], who propose to consider FTD-ALS as one of the focal presentations of ALS, alongside the bulbar and the limb-onset forms.
Genetics of FTD-ALS
Although clinical manifestations of a disease can be studied without any connection to the underlying pathology, the knowledge of pathologic changes often guides a physician to focus on specific clinical features and less so on others. It is conceivable, therefore, that much of the skepticism towards cognitive aspects of ALS stemmed from the fact that the first major genetic mutation found in this disease, SOD1, is associated with relatively well-preserved cognition [19]. For many years, SOD1-based models dominated ALS research. Thus, if the SOD1 phenotype was considered to be the classical manifestation of the disease, cognitive and behavioral changes must have looked like a rare and potentially irrelevant confound.
The discovery of the C9orf72 repeat expansion [13, 14] had exactly the opposite effect (see Chapter 14 for more detail). The fact that this mutation, much more common than SOD1, could be associated with FTD as well as ALS directed the attention of the research community to a possible overlap between both diseases. Research centers all over the world set out to examine whether patients with this mutation show any characteristic features which would distinguish them from those without the mutation. One of the most prominent was the already mentioned high prevalence of psychosis in C9orf72 patients [3]. However, it is important to remember that C9orf72 does not explain all familial cases of overlap between FTD, ALS, and neuropsychiatric disorders [82].
While the C9orf72 repeat expansion accounts for the largest percentage of FTD-ALS cases, other genes have also been implicated. A recent study describes a case of progressive aphasia associated with a mutation in the OPTN gene [83]. In contrast to the C9orf72 gene, OPTN gene seems to be more frequent in Japan than in Europe and the exact cognitive profile of patients with OPTN mutation is still to be determined. The differential distribution of the two genes raises, however, the question of to what extent the clinical phenotypes, including cognitive and behavioral features, might be influenced by the specific genetic background of the population in question: an issue to be addressed by future research.
Conclusions
The fields of MND and FTD are changing rapidly. Advances in molecular biology and genetics are illuminating the pathologic processes. The discovery of the C9orf72 gene mutation is likely to be followed by the identification of new genes and characteristic phenotypes associated with them. Of course, we hope that these discoveries are translated into new treatments as soon as possible.
And yet, underlying these breathtaking advances, there is a long tradition of related insights and observations. As pointed out in the introduction to this chapter, much of the current knowledge about the frequency, pattern, and natural history of cognitive and behavioral symptoms in ALS, including the idea of the ALS/FTD overlap, was observed before 1950. What has changed most dramatically are not the insights themselves, but the way in which they influence everyday clinical practice. The overlap between ALS and FTD is by now widely recognized by clinicians; cognitive assessment is becoming an integral part of the clinical evaluation of patients with MND, and cognitive status is likely to become an important variable in the evaluation of future pharmacologic trials. Thus, the relationship between ALS and FTD (and, in broader terms, between motor and cognitive functions in general [84]) has moved from a rare curiosity reported at the periphery of scientific developments into the very heart of current research and clinical practice.
References
Related posts:
 Practical management of frontotemporal dementia
The family’s perspective on FTD
Practical management of frontotemporal dementia
The family’s perspective on FTD
 Neuropathology of frontotemporal dementia and related disorders
Neuropathology of frontotemporal dementia and related disorders
 Functional disability and the impact of frontotemporal dementia in everyday life
Historical introduction to FTD
Functional disability and the impact of frontotemporal dementia in everyday life
Historical introduction to FTD
 Overview of frontotemporal dementia and its relationship to other neurodegenerative disorders
Overview of frontotemporal dementia and its relationship to other neurodegenerative disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


