The Generalized Epilepsies
Introduction
Epileptic syndromes with seizures arising simultaneously from both hemispheres have been broadly named generalized epilepsy. This group may be further divided into two others based on their presumed aetiology: idiopathic generalized epilepsies and cryptogenic or symptomatic generalized epilepsies. They have a respective annual incidence of 6.65 and 1.15 per 100 000, accounting for 40% of all epilepsies and making up a higher proportion of all paediatric epilepsies. There is often a great overlap in symptoms between the various syndromes, although the aetiologies vary over a broad spectrum. Table 2.1 lists the common clinical features of idiopathic generalized epilepsies. All of these conditions tend to be age specific with prominent genetic components that often are associated with channelopathy. The cryptogenic generalized epilepsies are also very similar in clinical presentation as well as having prominent genetic features. The epilepsy arising from the encephalopathic changes, however, often arises from structural malformations, hereditary or congenital disorders, or inborn errors of metabolism.
Table 2.1 Shared clinical features of idiopathic generalized epilepsies | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
Idiopathic generalized epilepsy (IGE) is a confusing term, referring to the fact there is no apparent structural cause and not meaning ‘of unknown cause’. IGE probably has a genetic basis of polygenic origin with variable penetrance. IGE accounts for 10-20% of all cases of epilepsy and can be subdivided as shown in Table 2.2.
Juvenile myoclonic epilepsy (JME) is the most common subtype of IGE, accounting for 10% of all (both adult and paediatric) epilepsies. Its incidence peaks between the ages of 12 and 18 years; however, it is often not recognized by the patient or family initially and is mistaken for morning clumsiness, as seizures usually occur upon awakening or
within a few hours of awakening. Seizures can be easily provoked by photostimulation in 5% of patients. Other clear precipitants include sleep deprivation, alcohol withdrawal and hypoglycaemia. The genetics of this disease are complicated and it is thought to have a polygenic inheritance. A positive family history is found in 25%, of which 5% are close relatives and, of those, approximately one-third have JME.
within a few hours of awakening. Seizures can be easily provoked by photostimulation in 5% of patients. Other clear precipitants include sleep deprivation, alcohol withdrawal and hypoglycaemia. The genetics of this disease are complicated and it is thought to have a polygenic inheritance. A positive family history is found in 25%, of which 5% are close relatives and, of those, approximately one-third have JME.
Table 2.2 Subdivisions of idiopathic generalized epilepsies | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
There are four syndromes that have been recognized by the International League Against Epilepsy (ILAE) whose seizures display ‘typical absences’: childhood absence epilepsy (CAE), juvenile absence epilepsy (JAE), epilepsy with myoclonic absences (EMA) and JME. JME will be discussed separately to this group due to the prominence of myoclonia in this syndrome. It should be noted that CAE, JAE and JME belong to IGEs, whereas EMA may be categorized under cryptogenic/sympatogenic generalized epilepsies.
CAE accounts for up to 10% of childhood epilepsy, peaking in incidence at 6-7 years and usually occurring in children without a learning disability or other neurological problem. Girls are twice as likely as boys to be affected and seizures may be readily induced through hyperventilation. The prognosis is good with rapid remission in up to 80% on treatment. By age 18 years, only approximately 20% of patients are still having seizures. JAE has a peak incidence at 10-12 years, although it can develop any time before 17 years of age. It has a very small preponderance for girls, though some authors would argue it has an equal incidence among the sexes. It has a high incidence of photosensitivity and is thought to be determined by multivariant genetics. It is probably a lifelong disorder and treatment should be continued lifelong despite absence seizures greatly decreasing in severity with increasing age. EMA, a rare epilepsy, has a peak age of onset at 7 years and a strong male preponderance. It shows little photosensitivity; however, seizures can be readily induced through hyperventilation. In contrast to the other absence syndromes, it has a less favourable prognosis due to the resistance of seizures to treatment, mental deterioration and the possible evolution to other types of epilepsy, including Lennox-Gestaut syndrome. It should be noted that almost half of the patients have intellectual and developmental disabilities before the development of absence seizures, and a further 25% go on to develop intellectual and developmental disabilities thereafter. There are other epileptic syndromes with absences, such as eyelid myoclonia with absences, perioral myoclonia with absences, photo- and pattern-induced absences and phantom absences with generalized tonic-clonic seizures (GTCS). They are, however, rare or due to their many similarities with other absence syndromes often misdiagnosed, highlighting the need for a new official classification of absence seizures.
Absence seizures are not an exclusive feature of generalized epilepsy. There is evidence that shows ‘typical absences’ may arise from focal pathology. Electroencephalography (EEG) studies have shown patients with subependymal heterotopias to experience ‘brief blank spells’ with simultaneous 3 Hz spike-wave discharges from the mesial surfaces of the frontal lobes.
GTCS may occur in many subtypes of IGE; however, epilepsy with grand mal on awakening (EGMA) is the only subtype that is characterized by this seizure type. As the name suggests, it is linked to sleep-wake cycles and occurs predominantly within the first hour of awakening and to a lesser extent in the evening when the patient relaxes after a busy day. Disturbances in the sleep-wake cycle as well as photostimulation can readily induce seizures. The age of onset ranges from 6 to 35 years with a peak in the second decade of life. It has a male preponderance and many patients have a positive family history for epilepsy, suggesting some genetic influence. EGMA may occur in conjunction with other subtypes of IGE, including CAE, JAE and JME.
Benign familial neonatal convulsions (BFNC) are one of the few syndromes where the genetics are understood and follow an autosomal dominant pattern of inheritance. It is twice as frequent in females as in males and usually seizures start about days 2-15 of life. Patients are usually full-term, otherwise healthy, neonates. The seizures usually remit within weeks, giving BFNC a good prognosis; however, 10% of patients subsequently develop epilepsy and another 5% develop febrile seizures. This syndrome is linked to a mutation in the voltage-gated potassium channel genes KCNQ2 and KCNQ3 creating reduced potassium conductance and increased neuron excitability. The remission may be explained by the upregulation of other potassium channels early in life to overcome this hyperexcitability. Benign myoclonic epilepsy of infancy (BMEI) is another epilepsy occurring in previously healthy infants, sometimes associated with a family history of epilepsy or febrile seizures. There is a preponderance for girls and the age of onset is from 4 months to 3 years. It has a good prognosis, responding well to treatment, which can be discontinued after 2-3 years. No developmental or neurological sequelae are observed, however, if treatment is started early.
It can be seen from Table 2.2 that there are other variants of IGE; however, they tend to well less defined, rarer and there are often overlaps with other generalized epilepsies, e.g. there is a considerable overlap between JME and EGMA. As the name suggests, myoclonic-astatic epilepsy has myoclonic seizures, as well as atonic seizures and is also associated with intellectual regression. 2.1 depicts the age of onset for the various divisions of IGE.
Cryptogenic or symptomatic generalized epilepsies are associated with diffuse brain dysfunction, which may be of known aetiology (i.e. symptomatic) or of unknown aetiology (i.e. cryptogenic). There is always clinical evidence of brain dysfunction, usually presenting through intellectual or motor developmental delay. The most important diseases in this group include West syndrome and Lennox-Gestaut syndrome. The two cardinal symptoms of West syndrome are infantile spasms with hypsarrhythmia and intellectual developmental delay. It reflects the immature brain’s response to a non-specific, generalized insult. It occurs only in the first year of life and previously gained visual and social skills are lost. The age of onset peaks at 4-6 months and it has an incidence of 1-2 per 4000 live births. There is also a positive family history in 7-17% of patients. Some of the common causes are listed in Table 2.3.
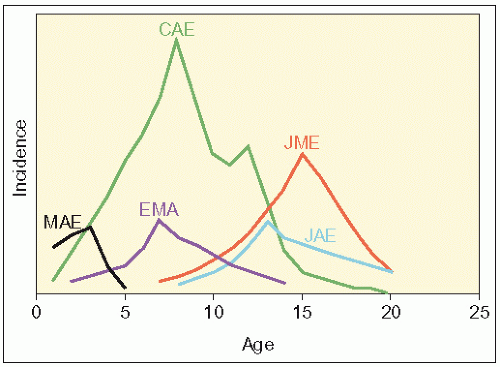 2.1 Relative incidence of human absence epilepsy syndromes during the first two decades of life. The five syndromes show developmental variation in the ages of seizure onset and duration of clinical seizure activity. In temporal order of peak incidence: MAE, myoclonic-astatic epilepsy; EMA, epilepsy with myoclonic absences; CAE, childhood absence epilepsy; JAE, juvenile absence epilepsy; JME, juvenile myoclonic epilepsy.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|





