CHAPTER 80 THE LEUKODYSTROPHIES
The leukodystrophies are now defined as disorders that (1) have a known or presumptive genetic cause; (2) have a progressive clinical course; (3) involve predominantly and usually confluently central nervous system (CNS) white matter; and (4) are characterized by a primary lesion of myelin or myelinating cells. This is the definition recommended by Powers1 and conforms to that proposed by Bielschowsky and Henneberg, who introduced this name in 1928.2 These diseases must be distinguished from abnormalities of myelin caused by infectious or toxic agents, disturbances of blood flow, anoxia or asphyxia, trauma, or genetic disorders in which damage to myelin or myelinating cells is a secondary event. The understanding, definition, and classification have been enhanced greatly by the combined use of neuroimaging and genetic techniques. Magnetic resonance imaging (MRI) is a powerful tool for the definition and categorization of leukoencephalopathies of unknown origin.3,4 For instance, MRI pattern analysis, combined with clinical data, led to the recognition that vanishing white matter disease5 (also referred to as childhood ataxia with diffuse central nervous hypomyelination)6 and megalencephalic leukoencephalopathy with subcortical cysts (MLC)7 are distinct clinical entities. Rapid application of positional cloning led to the identification of the gene defects.8–10 Although conventional MRI techniques have been of great value for the definition and classification of the leukodystrophies, these are now being supplemented by proton magnetic resonance spectroscopy (MRS), diffusion tensor imaging, and magnetization transfer techniques. One review summarized these novel techniques, as well as the current understanding of the alterations of myelin structure and function that underlie the neuroimaging abnormalities.11
Although these techniques represent important advances, van der Knaap cautioned against “diagnostic euphoria,”3 because for up to 50% of children with white matter abnormalities evident on MRI, no specific diagnosis could be established despite repeated MRI and extensive laboratory investigations.11 The 12 leukodystrophies in which the gene defect has been defined are the primary focus of this chapter.
X-LINKED ADRENOLEUKODYSTROPHY
Other names12 by which this disease is known include adrenomyeloneuropathy, Schilder’s disease, Addison’s disease with diffuse sclerosis, encephalitis periaxialis diffusa, Bronzekrankheit, and skelosierende encephalomyelitis.
Male Patients
Female Patients
Approximately 50% of women heterozygous for the adrenoleukodystrophy gene develop pure adrenomyeloneuropathy in middle age or later.16 The onset is later, and progression is slower than in male patients, but disability may be severe, necessitating the use of a wheelchair. Inflammatory brain disease17 and clinically or biochemically demonstrable adrenal insufficiency18 are rare in this disorder. They are present in about 1% of women heterozygous for the adrenoleukodystrophy gene.
Pathology
Inflammatory Cerebral Phenotype
In the inflammatory cerebral forms, there is loss of myelin, which is confluent and usually symmetrical with caudorostral progression.19 The periventricular and central white matter are involved. Cavitation and calcification may be seen. Arcuate fibers are relatively spared. In the most common form with posterior manifestation, the posterior cingulum, corpus callosum, fornix, hippocampal commissure, posterior limb of the internal capsule, and optic systems are typically involved. The cerebellar white matter exhibits similar but milder changes. In about 15% of affected patients, the cerebral pathology begins in the frontal white matter and advances rostrocaudally. Histopathological study reveals marked loss of myelin and, to a lesser degree, axons and a loss of oligodendrocytes in association with hypertrophic reactive astrocytosis. The pathology of the inflammatory cerebral lesions can be subdivided into three zones, which are also demonstrable in imaging studies.20 Zone 1, the advancing active edge of myelin loss, includes areas of intense perivascular inflammation and accumulation of lipid-laden macrophages. In Zone 2, there are large perivascular collections of mononuclear cells, particularly lymphocytes and mostly CD8 cytotoxic cells,21 which are highly characteristic in the area of early breakdown. In Zone 3, there are gliosis, loss of myelin, and variable loss of axons. In the inflammatory cerebral forms, there is also secondary corticospinal tract degeneration extending down through the peduncles, basis pontis, medullary pyramid, and spinal cord.
Pure Adrenomyeloneuropathy
The spinal cord bears the brunt of the disease process in men with pure adrenomyeloneuropathy and also in neurologically symptomatic women heterozygous for the adrenoleukodystrophy gene. Loss of myelinated axons and a milder loss of oligodendrocytes, but with little or no inflammatory response, are observed in the long ascending and descending tracts of the spinal cord.22 The pattern of fiber loss is consistent with a distal axonopathy in that the greatest losses are observed in the distal segments: that is, the cervical region for the ascending fasciculus gracilis and the low thoracic and lumbar segments for the descending corticospinal tract. The number of dorsal root ganglion cells is not reduced. This is indicative of axonal pathology and not neuronal damage as the primary event.23 Mitochondria are abnormal and contain lipid inclusions.24 Peripheral nerve lesions in adrenomyeloneuropathy are variable and mild in comparison with those of the myelopathy. Sural and peroneal nerves have displayed loss of large- and small-diameter myelinated fibers, endoneurial fibrosis, and thin myelin sheaths.25,26 Neurophysiological studies in 18 patients suggest that primary axonal pathology (present in 67%) is the principal abnormality. Only 9% of the patients fulfilled the electrodiagnostic criteria for primary demyelination.27
Adrenal Cortex and Testis
Adrenocortical cells become ballooned as a result of the accumulation of lamellae, lamellar lipid-laden cells, and fine lipid clefts. The striated material, which contains cholesterol esters esterified with very-long-chain fatty acids (VLCFAs), appears to lead to cell dysfunction, atrophy, and death.28 In fetuses affected by X-linked adrenoleukodystrophy (X-ALD), the fetal adrenal zone is already severely involved.29 In the testes, lamellae and lamellar lipid-laden cells are present in the interstitial cells of Leydig and their precursors. Degenerative changes in the seminiferous tubules and Sertoli cells are observed in adrenomyeloneuropathy30 and may eventually lead to azoospermia.31
Neuroimaging
Inflammatory Cerebral Phenotypes
The most common MRI abnormality in the childhood inflammatory phenotype is a pattern that involves the parieto-occipital white matter most severely. It is present in about 80% of affected patients.32–34 Arcuate fibers are relatively spared. After administration of gadolinium-diethylenetriamine pentaacetic acid contrast material, a rim enhancement can be seen surrounding the most severely affected area. Three zones can often be identified.32 The outer advancing zone is in the process of active demyelination without inflammation. Inflammation is present in the second zone. The third zone is completely demyelinated and gliotic, and cavitation and calcification may be present. Demyelination advances in the frontal direction. Structures that are affected relatively early are the lateral and medial geniculate bodies and, in some cases, the lateral-inferior part of the thalamus, the posterior limb of the internal capsule, and the external capsule. In the brainstem, there is involvement of the occipitoparietotemporopontine and pyramidal tracts, the brachia of the inferior colliculus and of the superior colliculus, and the lateral lemniscus. In approximately 15% of patients, the process starts in the frontal area with concomitant involvement of the rostrum and genu of the corpus callosum and of the anterior limb of the internal capsule,32,34 and then it spreads caudally. On occasion, the initial lesions are in the pons and cerebellum.35 The initial lesions may be asymmetrical and mistaken for a brain tumor.36
Proton MRS abnormalities precede changes demonstrable by conventional MRI37–39 and can be predictive of outcome; they aid in the selection of patients for hematopoietic cell transplantation (HCT) therapy and in the evaluation of therapies.40 Reduction of N-acetyl aspartate levels is the earliest change; increases in the choline peaks are correlated with the extent of the demyelinative process.
Adrenomyeloneuropathy
Conventional brain MRI usually appears normal in patients with “pure” adrenomyeloneuropathy. Abnormalities may be seen in the corticospinal and frontopontine tracts and have been interpreted as centripetal extension of the distal axonopathy.34,41 Brain MRS studies in patients with pure adrenomyeloneuropathy have revealed evidence of diffuse axonal pathology.42 Studies of the spinal cord with conventional MRI have demonstrated diffuse atrophy mainly in the lower thoracic cord.41 The magnetization transfer MRI appears to be a sensitive method for the quantitation of structural abnormalities in adrenomyeloneuropathy.43
Genetics
The mode of inheritance is usually classified as X-linked recessive, but the fact that 50% of heterozygous women are neurologically symptomatic indicates that a designation as X-linked is more accurate, as proposed by Dobyns and associates.44 The disease incidence is estimated at 1 per 17,000 and appears to be the same in all ethnic groups.45 The defective gene has been mapped to Xq28.46 The gene codes for a peroxisomal membrane protein, adrenoleukodystrophy protein (ALDP).47 ALDP is a member of the adenosine triphosphate-binding cassette (ABC) transporter superfamily.48 Forty-eight mammalian ABC transporters are estimated to exist. They transport a variety of substrates, including ions, sugars, amino acids, proteins, and lipids.49 Four mammalian peroxisomal ABC transporters have been identified and are designed as subgroup ABCD. The gene deficient in X-ALD has been assigned the name ABCD1. ABCD2 may also be relevant to X-ALD (see section on therapy). ABCD2, which has been mapped to 12q11, codes for the adrenoleukodystrophy-related protein ALDR, which has 66% homology with ALDP50 and can substitute for some of the functions of ALDP.51,52
More than 500 mutations in ALDP have been identified in patients with X-ALD53 and are updated in the X-linked Adrenoleukodystrophy Database website (www.x-ald.nl). Fifty-seven percent of the mutations are nonrecurrent, and many are unique to a kindred. Of all the mutations, 55.9% were found to be missense mutations, 27.1% nonsense mutations, 3.9% small in-frame amino acid insertions or deletions, and another 3.9% large deletions of one or more exons.53 No correlation between phenotype and genotype has been demonstrated. A single mutation may be associated with a wide range of phenotypes.54 It is common for widely varying phenotypes to co-occur in the same family. The action of a modifier gene has been postulated.55,56
Diagnosis
Definitive diagnosis of X-ALD depends on the demonstration of biochemical abnormalities and on results of mutation analysis. Demonstration of characteristic abnormalities in the levels of saturated VLCFAs in plasma is the most frequently used test.57 Statistically significant increases in the levels of hexacosanoic acid (C26:0) and in the ratios of C26:0 to C24:0 and to C22:0 are present in more than 99% of male patients with adrenoleukodystrophy and in approximately 85% of women heterozygous for the X-ALD gene.58 Increases in VLCFA levels in patients with other peroxisomal disorders, such as Zellweger’s syndrome, neonatal adrenoleukodystrophy, and infantile Refsum’s disease,59 can usually be distinguished easily on the basis of clinical manifestation and tests of the other peroxisomal functions. Patients on the ketogenic diet may produce false-positive results60; VLCFA levels are also increased in cultured skin fibroblast,61 and study of cultured fibroblasts can be helpful when results of plasma samples are equivocal. VLCFA levels are also increased in red blood cell membranes,16,62 in leukocytes,63 and in cultured amniocytes64 and chorionic villus cells.65
The plasma VLCFA assay57 is highly reliable for the identification of affected male patients. Abnormalities are already present on the day of birth.57 There are two reports of false-negative test results in a total of three male patients with X-ALD,66,67 but this has not observed in more than 100 affected male patients.57 In contrast, false-negative results occur in 15% to 20% of women heterozygous for the X-ALD gene. Because of this, mutation analysis is much preferred for the determination of carrier status in women at risk for X-ALD. More than 500 pathogenic mutations in the ABCD2 gene have been identified in patients with X-ALD53 and are updated in the X-linked Adrenoleukodystrophy Database website. Reliable methods for their identification in affected men and in heterozygous women have been published.68 The first step is to define the nature of the mutation in affected male family members or in a family member who is an obligate carrier. Once the mutation is found in a nuclear family member or a more distant relative, it can be determined whether this mutation exists in lymphoblasts or cultured fibroblasts of the at-risk person. VLCFA analysis in cultured amniocytes or chorion villus samples is a valuable technique for the identification of affected fetuses.65 However, false-negative results have been reported,69,70 and confirmation by mutation analysis should be performed when the nature of the mutation has been defined in an affected male patient or obligate heterozygote family member.
Pathogenesis
Role and Pathogenesis of Very-Long-Chain Fatty Acid Excess
The abnormal accumulation of saturated VLCFAs is the principal biochemical abnormality in X-ALD.14,71 There is considerable evidence that this contributes to pathogenesis, according to studies in model membranes72 and in cultured human adrenal cells73 and correlative studies in postmortem brain74 and adrenal tissue.75 Excess saturated VLCFAs are normally oxidized in the peroxisome.76 This process is impaired in cultured skin fibroblasts, leukocytes, and amniocytes of patients with X-ALD.77 The defect in X-ALD was localized to the first step: namely, the formation of the coenzyme ester of VLCFA acids.78,79 This reaction is catalyzed by VLCFA-coenzyme A synthetase (VLCS).80 VLCS structure and levels are normal in patients with X-ALD and in the X-ALD animal model.81–83 ALDP, the protein that is deficient in X-ALD, is required for the oxidation of VLCFA in fibroblasts of patients with X-ALD.51,52,84 ALDP has no homology with VLCS; as already noted, it is a member of the ABC transporter superfamily.48 The mechanism by which ALDP influences VLCFA metabolism is complex and not yet understood.85 Adding to the complexity are findings of several earlier86 and more recent studies87 that suggest that VLCFA synthesis is also increased in X-ALD.
Pathogenesis of Pure Adrenomyeloneuropathy
The studies of Powers and associates established that the primary defect in pure adrenomyeloneuropathy is a noninflammatory axonopathy that affects most severely the distal portions of the dorsal columns and corticospinal tracts in the spinal cord.22,23 The demyelination appears to be secondary to the axonal damage. The mechanism responsible for the axonal dysfunction has not been defined. It is postulated that the VLCFA excess destabilizes the axonal membrane or impairs the function of trophic factors.23 Mitochondrial abnormalities23 and oxidative stress88 have been proposed as contributing to pathogenesis. The pathology and clinical features in the mouse model of X-ALD resemble those of adrenomyeloneuropathy,89 and further studies in this model may increase understanding of the pathogenesis of adrenomyeloneuropathy.
Pathogenesis of Inflammatory Cerebral Forms of X-Linked Adrenoleukodystrophy
The most generally accepted concept for the pathogenesis of the inflammatory response is that the accumulation of VLCFA has an adverse effect on myelin and oligodendrocyte stability and function, which renders them vulnerable to various other adverse events (a “second hit”), which in turn initiates a destruction cascade that results in the death of oligodendrocytes and rapid destruction of myelin.90 Cytokines, such as tumor necrosis factor α, play an important role.91 Apoptosis of oligodendrocyte has been reported.92 Ito and colleagues21 demonstrated that most of the perivascular cells that accumulate in the active demyelinating lesions are CD8 cytotoxic cells. They concluded that oligodendrocyte death was lytic/cytotoxic rather than apoptotic. There was strong CD1 immunoreactivity in astrocytes and microglia. CD1 molecules are antigen-presenting glycoproteins that, unlike the major histocompatibility complex proteins, can present self-lipid antigens to T-cells.93 This has led to the intriguing hypothesis that VLCFA-containing complex lipids, which are present in brain tissue from patients with X-ALD but not in normal brain tissue, act as triggers for the autoimmune response in patients with the inflammatory adrenoleukodystrophy phenotype. Asheuer and associates94 demonstrated decreased expressions of an ABCD transporter gene (ABCD4) and a VLCS (BG1) in patients with X-ALD and their susceptibility to the inflammatory phenotype. This is the first demonstration of a possible genotype-phenotype correlation in X-ALD.
Therapy
Steroid Replacement Therapy
The importance of evaluating adrenal function and providing appropriate replacement therapy cannot be overemphasized. Steroid replacement therapy in general does not alter neurological progression, except possibly in some patients with adrenomyeloneuropathy,95 but it can improve strength and well-being and may be lifesaving. Most male patients with X-ALD have increased plasma adrenocortical hormone and impairment of cortisol responsiveness to a 0.25-mg intravenous dose96 of cosyntropic (Cotrosyn) after 60 minutes. The authors and colleagues recommend that at least one of these tests be performed yearly. Isolated measurements of plasma cortisol levels are insufficient and may lead to the false conclusion that adrenal insufficiency has been ruled out. Glucocorticoid dose requirements are generally the same as those used for other forms of primary adrenal insufficiency.97 To mimic diurnal rhythms of physiological cortisol secretion, adult patients receive 25 mg of cortisone acetate or 20 mg of hydrocortisone in the early morning and a smaller second dose, 12.5 or 10 mg, respectively, in the late afternoon. The dosage in children is 5 to 10 mg per 24 hours. Not all require mineralocorticoid replacement. When postural hypotension, hyponatremia, or hyperkalemia does persist despite adequate glucocorticoid therapy, fludrocortisone, 0.05 mg to 0.1 mg/day, is prescribed.
Hematopoietic Cell Transplantation
HCT is an important therapeutic modality for the cerebral inflammatory forms of X-ALD. Either bone marrow cells or umbilical cord cells are used.98 Aubourg and coauthors99 were the first to report stabilization and reversal of neurological manifestation in an 8-year-old boy who had early evidence of cerebral involvement. This patient was healthy 10 years later (P. Aubourg, personal communication, July 2000). Shapiro and associates100 reported long-term stabilization in patients who had received bone marrow cells. Peters and colleagues98 provided follow-up on 126 patients with X-ALD who received HCT. This study provided important guidelines in regard to the selection of patients for HCT. The overall 5-year survival rate was 56%, compared with an estimated 45% rate among untreated patients. The rate of transplantation-related mortality was 14%. Outcome with respect to mortality, morbidity, and quality of life was unsatisfactory for patients who were already severely ill at the time of HCT, and the procedure is not recommended for them. In contrast, the 5-year survival rate was 92% in a group of patients whose neurological involvement (performance IQ > 80) and neuroradiological involvement (Loes MRI score < 9, range = 0-34)101 were still relatively mild, and the procedure is recommended for them. Peters and colleagues98 provided a more detailed discussion of the indications for HCT. At this time, the procedure is not recommended for asymptomatic patients with normal MRI findings or for those with pure adrenomyeloneuropathy. The mechanism of the beneficial effect of bone marrow cell transplantation is incompletely understood.14
Dietary Therapy with Lorenzo’s Oil
Lorenzo’s oil is a 4:1 mixture of glyceryl trioleate and glyceryl trierucate. It has the remarkable biochemical effect of normalizing the plasma levels of VLCFA in patients with X-ALD within 4 weeks, probably by inhibiting endogenous VLCFA synthesis.102 Investigators reported that administration of Lorenzo’s oil to asymptomatic boys who had normal brain MRI findings significantly reduced their risk of developing childhood cerebral X-ALD.103 It was recommended that asymptomatic patients with normal brain MRI findings who are younger than 8 years be placed on a carefully supervised program of Lorenzo’s oil and dietary therapy. Adrenal function and brain MRI appearance must be monitored at 6-month intervals. Lorenzo’s oil does not alter significantly the neurological progression in patients with X-ALD who already have evidence of inflammatory cerebral involvement104,105 and in adrenomyeloneuropathy.106 However, results of a single cohort study suggested that it may slow the progression of adrenomyeloneuropathy in patients who did not have evidence of cerebral involvement.107 A double-blind placebo-controlled study of Lorenzo’s oil therapy in men and women with pure adrenomyeloneuropathy is now in progress.
Other pharmacological therapeutic approaches have shown therapeutic potential in preclinical studies and preliminary clinical trials. These include 4-phenylbutyrate,51 lovastatin,108,109 and lovastatin with arginine butyrate.110
Gene replacement therapy is under active investigation. Preclinical studies have shown encouraging results.111,112
METACHROMATIC LEUKODYSTROPHY
Clinical Manifestations
There are late infantile, juvenile, and adult manifestations. In about 75% of patients, the late infantile and juvenile forms are equally divided in frequency. The other 25% have the adult form.113
Late Infantile Form
This form manifests between 6 months and 4 years of age and has been subdivided into four clinical stages.114 In clinical stage I, there is hypotonia of the legs or of all four limbs. If the child is walking, the gait becomes unsteady. Deep tendons are diminished or absent. This stage lasts for a few months to more than a year. In stage II, the child can no longer stand and shows mental regression. Speech deteriorates as a result of dysarthria and aphasia. Nystagmus, ataxia, and truncal signs develop, and muscle tone in the legs is increased. This stage lasts only a few months. In stage III, the flaccid paresis is superseded by spastic tetraplegia with pathological reflexes and extensor plantar reflexes, and the child becomes bedridden. Feeding difficulties and episodes of airway obstruction occur. About 25% of patients develop epileptic seizures. In stage IV, the patients enter a decerebrate state and lose all contact with their surroundings. Death usually occurs about 5 years after onset of clinical symptoms.
Adult Form
Onset of adult MLD has been reported at varying ages up to age 63.115 Patients show gradual decline in intellectual abilities. They become emotionally labile and show memory deficits, disorganized thinking, behavioral abnormalities, or psychiatric symptoms such as hallucinations or delusions.116 Clumsiness of movement and urinary and sometimes fecal incontinence are also present. A progressive spastic paresis of the arms and legs develops, with increased tendon reflexes and extensor plantar reflexes. Ataxia and extrapyramidal symptoms and dystonia may be present, as well as optic atrophy and signs of bulbar dysfunction. Peripheral neuropathy is often absent.117
Pathology
The pathology of MLD in the nervous system is characterized primarily by demyelination and deposits of metachromatic granules in the central and peripheral nervous systems. High-resolution electron microscopy of the storage granules revealed that the inclusions are surrounded by a membrane, which suggests that they are located in the lysosome.115 The central white matter is reduced in amount, is firm, and in severely affected regions may show cavitation of spongy degeneration. The subarcuate fibers are usually spared. There is moderate to severe loss of myelin sheaths. The intrafascicular oligodendrocytes are reduced in number. There is a striking accumulation of metachromatic granules in macrophages that are prominent in perivascular spaces and also in oligodendrocytes and in neurons. Immunocytochemical studies have shown that the accumulated material in neurons and glial cells contains sulfatide.118 The cerebellum is atrophic with severe demyelination, prominent gliosis, storage granules, and a reduction of Purkinje cells. The peripheral nervous system shows segmental demyelination. Metachromatic granules are present in Schwann cells and in endoneurial macrophages.
Neuroimaging
MRI reveals periventricular white matter abnormalities with a symmetrical distribution.119 In juvenile and adult cases, there is preferential frontal lobe involvement. The arcuate fibers are relatively spared. The corpus callosum is invariably affected. The posterior limb of the internal may be involved. Brainstem lesions in the pyramidal tract may be observed, and the cerebellar white matter may be involved. Contrast enhancement is usually absent; this helps differentiate MLD from X-ALD, in which enhancement is often prominent. Diffusion-weighted images vary with the stage of the illness. Oguz and associates reported that diffusion was restricted in the early stage and increased in the later stage.120
MRI in MLD often exhibits radially oriented hypointense stripes in T2-weighted hyperintense areas. Postmortem studies in which neuroimaging patterns were correlated precisely with histopathological findings revealed that the stripes corresponded to perivenular zones in which there was relative preservation of myelin, together with the accumulation of lipid-laden glial cells.121
Genetics
The mode of inheritance is autosomal recessive. The incidence of late infantile MLD varies in different countries.115 MLD appears to be most common in northern Sweden, where the incidence is estimated to be 1 per 40,000 among some groups, and among Arabs living in Israel. In France and in Germany, the incidence was estimated to be 1 per 130,000 to 1 per 170,000.
The defective gene has been mapped to locus 22q13. It codes for arylsulfatase A, which catalyzes the desulfation of 3-sulfogalactosyl-containing glycolipids. Four such glycolipids have been identified.115 Of these, galactosylceramide-3-sulfate is present in the highest concentration and most relevant to MLD.
Metachromatic Leukodystrophy-Causing Mutations
Sixty-three MLD-causing mutations have been identified. The mutations can be subdivided into two general categories: the null alleles, in which the mutation does not allow synthesis of any function, and the R-alleles, in which there are low amounts of residual enzyme activity. Null alleles tend to be associated with the late infantile phenotype, R-alleles with the adult phenotype, and heterozygosity for null and R-alleles with the juvenile phenotype. However, there is considerable phenotypical variability of severity when patients with the identical genotype are compared.122
Saposin B Deficiency
A small number of patients with clinical features that resemble those of MLD have a deficiency of saposin B, the sulfatide activator protein. These patients have increased levels of sulfatide in tissues and urine, but arylsulfatase A activity is normal. Nine cases have been reported and include patients with late infantile, juvenile, and adult onset.123 The molecular defects involve homoallelic point mutations that destroy a glycosylation site.
Multiple Sulfatase Deficiency
In multiple sulfatase deficiency, the catalytic activity of 12 sulfatases, including arylsulfatase A, is impaired. More than 50 cases have been described.124 The clinical manifestation resembles that of classic MLD but with certain additional features such as mildly coarse features, dysostosis multiplex, stiff joints, ichthyosis, hydrocephalus, deafness, and enlarged liver, attributable to defects in one of the other sulfatases. The gene defect leads to reduced activity of a post-translational system that generates an α-formyl glycine residue from a thiol group of an active site cysteine that is conserved in all members of the mammalian sulfatase family.
Diagnosis
The laboratory diagnosis of MLD depends on the demonstration of decreased arylsulfatase A activity and increased excretion of sulfatide in urine. The arylsulfatase A assay is performed in peripheral blood leukocytes or cultured fibroblasts.125 The major problems in the enzymatic diagnosis is the frequent occurrence of the clinically benign pseudodeficiency alleles, which reduce arylsulfatase A activity. Measurement of urinary sulfatide excretion is key to the distinction between MLD and pseudodeficiency. This excretion is markedly increased in patients with MLD and normal in patients with pseudodeficiency or in persons heterozygous for the MLD-causing mutation.126 MLD and pseudodeficiency can also be distinguished by the sulfatide loading assay.127 Patients with multiple sulfatase are distinguished by demonstration of deficient activity of arylsulfatase B and C, in addition to the defect of arylsulfatase A. Patients with saposin B deficiency have normal arylsulfatase A activity, but their sulfatide loading value and urinary sulfatide excretion are increased. Mutation analysis is of great value for the identification of persons heterozygous for the MLD-causing mutation. It detects the pseudodeficiency. However, it must be kept in mind that alleles bearing the relatively common pseudodeficiency polymorphism may, in addition, contain an MLD-causing mutation.
Pathogenesis
The accumulation of sulfatide, secondary to the deficiency arylsulfatase A, is the principal biochemical abnormality in MLD. The sulfatides accumulate mainly within the lysosome. The sulfatide accumulation is noted in several extraneural tissues, such as the gallbladder, liver, kidneys, pancreas, adrenal cortex, and sweat glands, but it leads to pathological changes only in the gallbladder.
The demyelination that occurs in MLD appears to be secondary to sulfatide-induced changes within the cells responsible for myelin maintenance: namely, the oligodendrocytes in the CNS and the Schwann cells in the peripheral nervous system.129 Changes in the subcellular organelles of these cells are observed before any morphological abnormalities in the myelin sheaths associated with them are detectable.115 Investigators who used immunocytochemical techniques have demonstrated the accumulation of sulfatides in neurons and glial cells in arylsulfatase A-deficient mice.118 The degeneration of neurons and glia is enhanced and accelerated by the secretion of proinflammatory cytokines by monocytes.130
Other pathogenetic mechanisms that may contribute are impaired myelin stability secondary to its abnormal biochemical composition131 and the abnormal accumulation of sulfogalactosylsphingosine (lysosulfatide).132 Lysosulfatide has been shown to inhibit the activity of protein kinase C and cytochrome oxidase at concentrations far below those found in tissues of patients with MLD.132,133
Therapy
Bone marrow transplantation is under investigation as a therapy for MLD. It has not been effective in symptomatic patients with the late infantile form of the disease and may even accelerate the progression of the disease.134 Several reports suggest that bone marrow transplantation arrests the progression in patients with the juvenile or adultonset MLD phenotype.135–137 Evaluation of these results is difficult because of the relatively short follow-up and the variability of progression in untreated patients. Several presymptomatic patients have undergone transplantation.138,139 Longer follow-up is necessary to assess effectiveness.
Therapeutic studies in the mouse model of MLD are highly encouraging. Ex vivo administration of genetic modification of hematopoietic stem cells led to a remarkable correction of neuropathological changes.140 Matzner and coauthors reported the unexpected and surprising findings that intravenous administration of purified arylsulfatase A improved nervous system pathology and function.141
GLOBOID LEUKODYSTROPHY
Clinical Features
The infantile form of the disease is most common. Hagberg and associates subdivided the course of the disease into three stages.142 In stage I, the child, apparently normal during the first few months after birth, becomes hyperirritable and hypersensitive to auditory and tactile stimuli, and there is some stiffness of the limbs. Slight regression of psychomotor development, feeding difficulties, and vomiting may be observed. Early peripheral nervous system manifestations are common.143 In stage II, there is rapid deterioration. There is marked hypertonicity, with extended and crossed legs, flexed arms, and arched back. Optic atrophy and sluggish pupillary reflexes are common. Stage III is the “burnt-out” stage, sometimes reached within few weeks or months. The child is in a decerebrate state, is blind, and has no contact with his or her surroundings. Patients rarely survive for more than 2 years.
Juvenile- and adultonset cases are well documented. The juvenile cases have been subdivided into the late infantile (early childhood)-onset form, which begins at ages 6 months to 3 years, and the late childhood-onset form, which begins at ages 3 to 8 years.144 The early childhood form progresses rapidly, with death approximately 2 years after onset. The late childhood cases manifest with loss of vision and with hemiparesis, ataxia, and psychomotor regression; death occurs 10 months to 7 years after onset. The number of reports of adultonset cases is increasing. Progressive paraparesis and tetraparesis with demyelination in the spinal cord are often the main abnormalities. One female patient developed slowly progressive paraparesis at 38 years of age.145 Other cases were misdiagnosed as amyotrophic lateral sclerosis.146
Pathology
The pathology is confined mainly to the nervous system.147 Postmortem examination reveals that the brain is small and atrophic, with shrunken gyri and widened sulci. The major histopathological changes are demyelination, gliosis, and the presence of unique macrophages, the globoid cells in the white matter. The subarcuate fibers tend to be spared. The phylogenetically newer fiber tracts are usually more involved in the demyelinating process. In the spinal cord, the pyramidal tracts are more affected than are the dorsal columns. The oligodendroglial cell population is severely diminished in the areas of demyelination. Globoid cells are clustered around blood vessels and may be multinucleated. They contain tubular inclusions that have morphological similarities to the inclusions of pure galactosylceramide.148 Secondary axonal degeneration is a consistent finding. The peripheral nerves are commonly affected. They show endoneurial fibrosis, proliferation of fibroblasts, and segmental demyelination. Inclusions similar to those in the globoid cells in the brain are found in the cytoplasm of histiocytes, macrophages, and Schwann cells.
Neuroimaging
In the early stages, computed tomographic changes may be more evident than MRI changes. In stage I, computed tomographic scans may be normal or exhibit symmetrical increased density in the thalami, the corona radiata, and the posterior limb of the internal capsule and basal ganglia.149 MRI confirms the presence of white matter abnormalities with relative sparing of arcuate fibers in infantile-onset GLD (Fig. 80-1). The posterior limb of the internal capsule, corpus callosum, cerebellar white matter, and brain tracts are also involved. In cases of adultonset GLD, both computed tomography and MRI reveal predominant involvement of the occipital periventricular white matter with extension in the parietal and temporal direction and associated involvement of the splenium of the corpus callosum, a pattern that resembles that seen in X-ALD. In adultonset cases, symmetrical high-signal intensity lesions may be evident on T2-weighted MRI in frontoparietal white matter, the centrum semiovale, and the posterior limb of the internal capsule, with sparing of the periventricular white matter (see Fig. 80-1).145 MRS in patients with infantile-onset GLD revealed pronounced elevation of myoinositol- and choline-containing compounds, which reflected gliosis and demyelination. N-acetyl aspartate levels were decreased, which reflected neuroaxonal loss.150
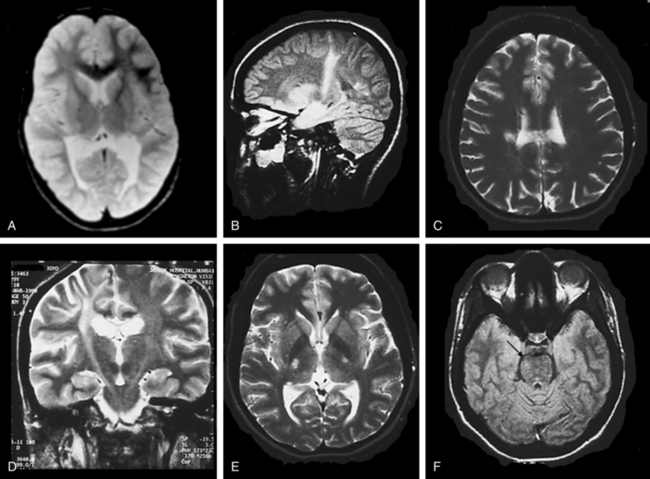
Genetics
The mode of inheritance is autosomal recessive. The gene has been mapped to loci 14q24.3-32.1. It codes for galactocerebroside (psychosine) β-galactosidase. The incidence is estimated to be 1 per 100,000 in the United States but appears to be higher in the Scandinavian countries. More than 60 mutations have been identified.147,151 Although there is no consistent correlation between genotype and phenotype,152 some associations have been identified. The 502T/del mutation accounts for 50% of mutant alleles in The Netherlands and 75% in Scandinavian countries. The mutation probably occurred initially in Sweden and traveled from there throughout Europe, Asia, and the United States. This 30-kilobase deletion eliminates all of the coding region of one of the subunits of the enzyme, and patients who are homozygous for this deletion have the severe infantile-onset phenotype. Two other mutations, C1538T and A1652T, account for about 10% to 15% of mutant alleles of infant patients with European ancestry. The G809A mutation is common in patients with adultonset GLD. One copy of the G809A mutation is probably enough to explain a mild phenotype in heterozygous patients whose other allele has a mutation that usually is associated with a severe phenotype.147 A series of polymorphic changes in the gene that occur on the same allele as disease-causing mutations have been identified; they influence the enzyme activity and clinical manifestations in persons who are heterozygous for GLD-causing mutations.153
Diagnosis
Assays of GalC activity in peripheral leukocytes or cultured fibroblasts is the most reliable method for diagnosis.154 Leukocyte and cultured fibroblast results are equally reliable. Wenger and colleagues147 listed citations for validated techniques that use natural substrates. The same assay in cultured amniotic cells or in biopsy specimens of chorionic villi is used for prenatal diagnosis, but it is valuable for measuring the GalC enzyme activity in the carrier parents beforehand, because some carriers have enzyme levels that are sufficiently low to characterize them as homozygous affected. When this is the case, special care must be taken in the interpretation of the results of the prenatal study, and the results can be complemented by mutation analysis. The wide range of enzyme activity in normal individuals and in carriers, which results mainly from polymorphic amino acid changes that cause a wide range in the values tested, limits the reliability of the enzyme assay for carrier identification. In families in which the mutations have been defined, carrier status is determined by mutation analysis. An important diagnostic development is the progress that has been made toward mass neonatal screening for lysosomal disorders, including GLD,155–157 Neurophysiological studies, such as motor conduction velocity and visual and auditory evoked responses are abnormal in most patients with GLD,158 but it is important to realize that they may be normal in patients with the adultonset phenotypes.159
Pathogenesis
The globoid cell, the hallmark pathological feature of GLD, is the consequence of the accumulation of galactosylceramide. Galactosylceramide is unique among sphingolipids in its ability to elicit the globoid cell reaction when injected into normal rat brain.160 Even though the galactosylceramide thus plays a role in some aspects of the pathogenesis of GLD, there is increasingly compelling evidence that it is the accumulation of psychosine (sphingosine-galactose) that is the principal pathogenetic factor. This pathogenetic mechanism, now referred to as the psychosine hypothesis, was first proposed by Miyatake and Suzuki161 and reexamined in detail 25 years later by Suzuki.162 GalC, the gene product that is deficient in GLD, catalyzes the degradation of both galactosylceramide and psychosine. Psychosine is present in very low concentration in normal brain tissue, but its concentration in GLD white matter is increased 100-fold.147
Psychosine with its free amino group is highly cytotoxic. Its effect is mediated by caspase activation.163 Psychosine-induced apoptosis is a mouse oligodendrocyte precursor line mediated by caspase activity. Oligodendrocytes are selectively destroyed because psychosine formation occurs primarily in these cells. The molecular mechanisms of psychosine-induced cell death have been clarified.164 Psychosine-induced apoptosis in human oligodendrocyte cell line studies in animal models, particularly the canine model and the twitcher mouse, have been of great value for the studies of pathogenesis of GLD and its therapy.165
Therapy
HCT and bone marrow transplantation for GLD are under intense investigation. HCT was of benefit in four patients with adultonset GLD: CNS deterioration was reversed, MRI findings improved in three patients, and levels in cerebrospinal fluid (CSF) diminished.166 The effect of HCT in patients with infantile-onset GLD and in asymptomatic infants with GLD has been evaluated.167 Although the symptomatic boys with infantile GLD experienced no benefit, there was strong evidence of a beneficial effect in 11 asymptomatic newborns who received umbilical cord blood transplants at 12 to 44 days after birth. At the time of follow-up (median age, 3.0 years) they had age-appropriate cognitive function and receptive language skills, and serial MRI studies demonstrated progressive central myelination. There is no doubt the procedure has a profound effect with regard to mortality and early development, but there were mild-to-moderate delays in expressive language and gross motor function. Additional follow-up is needed to assess the long-range outcome. Ethical aspects of this therapeutic approach were discussed in an editorial.168
Another approach, therapy aimed to reduce the concentrations of substrate, is being tested in experimental animals.169 Studies of transplantation of neural cells and direct infection of the brain with viral vector containing GalC, are in progress in experimental animals.
PELIZAEUS-MERZBACHER DISEASE
Pelizaeus-Merzbacher disease is known as several entities: classic Pelizaeus-Merzbacher disease,170 connatal Pelizaeus-Merzbacher disease,171,172 X-linked spastic paraparesis type 2 (SPG2),173,174 proteolipid protein (PLP) null syndrome, and pure spastic paraplegia.
Clinical Features
Classic Pelizaeus-Merzbacher Disease
This disorder is characterized by abnormal eye movements (horizontal or rotatory) with onset during the first months of life; psychomotor deterioration before 2 years of age; and the appearance of bilateral pyramidal tract signs, dystonia, and often ataxia during the first years of life. Laryngeal stridor and optic atrophy are frequent.170
Connatal Pelizaeus-Merzbacher Disease
This disorder is characterized by nystagmus at birth, pharyngeal weakness, stridor, hypotonia, severe spasticity, and seizures.174
Complicated Spastic Paraplegia
This disorder is characterized by nystagmus, ataxia, and spastic gait, but little or no cognitive impairment.174
Pathology
Features include lack of myelin in all parts of the CNS. Islets of preserved myelin around blood vessels in deeper parts of brain produce the so-called tigroid pattern. Axons are relatively preserved. Oligodendrocytes are reduced in number, as is the number of cytoplasmic processes. A length-dependent axonal degeneration is also present.173
Neuroimaging
MRI reveals an arrest of myelination. In some cases, no myelin appears to be present; in other cases, myelin is present in parts of the brainstem, cerebellar white matter, the posterior limb of the internal capsule, the thalamus, and the globus pallidus. The pattern described is normal for a neonate or an infant in the first few months of life, although not normal at the age of the patient. The white matter may appear speckled, reflecting the tigroid pattern seen pathologically. There also is evidence of neuroaxonal injury, based on the demonstration of significant and widespread reduction in brain N-acetyl aspartate levels.176
Genetics
The mode of inheritance is X-linked recessive. Some women heterozygous for Pelizaeus-Merzbacher disease develop neurological progressive paraparesis.175,177,178 The gene maps to locus Xq22 and codes for PLP. This protein makes up approximately 50% of myelin protein weight. It is composed of four helices that span the cell membrane. Portions of the protein chain extend into the extracellular space, where they have a homophilic interaction with PLP molecules in adjacent spirals of the cell membrane.179 This region is visualized as the intraperiod line on electron microscopy.180
Duplication of the region surrounding locus Xq22 accounts for the majority, perhaps up to 70%, of the mutations in PLP.181 These duplications arise from sister chromatid exchange. Striking variations in the breakpoints and in the size of the duplication occur.182,183 Most patients with mutations have the classic phenotype182; some have the connatal form,184 and others have the mild SPG2 phenotype. Deletions of the PLP locus occur only rarely.185,186 Patients in whom no PLP protein product is formed, referred to as null mutations, tend to have a mild phenotype with relatively mild spastic paraparesis, which progressed during adolescence and (unlike that in classic Pelizaeus-Merzbacher disease) was associated with demyelinating peripheral neuropathy.173 About 20% of patients have point mutations at the PLP locus that alter the amino acid sequence of the PLP/DM20 proteins. Approximately 100 distinct mutations have been discovered to date. The majority of abnormal PLP/DM20 proteins result in severe phenotypes through a toxic gain of function, whereas the remainder result in a milder form that is associated with loss of function.
Diagnosis
Although the classic phenotype has a relatively distinct clinical manifestation,170 the connatal form can be confused with motor neuron disease or spinal muscular atrophy. The mildest forms merge clinically with the syndromes of SPG2. MRI is essential for the evaluation of individuals with clinical signs and symptoms of classic Pelizaeus-Merzbacher disease or SPG2. Virtually all patients with Pelizaeus-Merzbacher disease eventually have MRI findings consistent with a leukodystrophy.187,188
A search for proteolipid protein 1 (PLP1) gene duplication is the most efficient initial screening test. Both interphase fluorescent in situ hybridization and quantitative polymerase chain reaction should be used.182 If neither method yields a positive result, direct sequencing of the PLP1 gene should be performed.189 Prenatal diagnosis has been accomplished.190
Pathogenesis
Because mutations in which no PLP1 protein is synthesized lead to the mildest disease, it has been postulated that toxic effects are caused either by overexpression of the gene in patients with duplications of the normal gene or by the presence of mutant genes. In patients with the gene duplications, excessive amounts of PLP1 have been shown to accumulate in the late endosomal and lysosomal compartments. PLP1 normally is associated with cholesterol and other lipids to form “lipid rafts” that traffic through the Golgi compartment. In rodent models of Pelizaeus-Merzbacher disease in which there is an excess of PLP1, these lipid rafts are shunted to the late endosomal and lysosomal compartments, effectively draining myelin lipids from the Golgi compartment.191 In patients with mutations in the PLP1 coding region, the differences in clinical severity appear to be related to differential effects of the mutation on protein folding and trafficking.192 These unfolded proteins accumulate in the endoplasmic reticulum, and this leads to the unfolded protein response.193,194 This response, which involves the increased expression of unfolded protein response effector genes, protects the cell against metabolic stress to preserve or reestablish homeostasis. In cells in which homeostasis cannot be maintained, there often is apoptosis by activation of caspase cascades.195 This pathogenetic mechanism is presumed to be operative in oligodendrocytes and neurons. The understanding of the pathogenesis of Pelizaeus-Merzbacher disease has been aided greatly by studies in animal models of the disease.196 These include the “jimpy” mouse and the “jimpy-msd” mouse, in which the disorder resembles classic Pelizaeus-Merzbacher disease; the “jimpy-4j” mouse, in which the disorder resembles connatal Pelizaeus-Merzbacher disease; and the “rumpshaker” mouse, which is a model of SPG2. The myelin-deficient rat also provides a model of connatal Pelizaeus-Merzbacher disease. Because rats are larger animals, this model is well suited for neurophysiological studies of pathogenesis and for the study of therapeutic interventions.197,198
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


