Fig. 10.1
Post mortem human brain at the level of the caudate-putamen. Coronal brain sections taken through the caudate-putamen of a normal (left) and a Huntington’s disease patient (right). The Huntington’s disease brain on the right shows degeneration of the caudate nucleus adjacent to the lateral ventricle, which has enlarged in response to the striatal atrophy. (Courtesy of J-P. Vonsattel. Reproduced from Alexi et al. (2000), with permission from Elsevier)
Like many other neurodegenerative diseases , another hallmark of HD is the progressive aggregation or inclusion body formation of mutant HTT (mHTT). These aggregates/inclusions were initially identified in a mouse model harboring an expanding CAG repeat stretch within exon1 of a human HTT transgene (Davies et al. 1997) . Subsequently, these inclusions were identified in the neurons of HD patients (DiFiglia et al. 1997) . The initial descriptions of these inclusions in HD tissue, largely identified them as neuronal in nature, cytoplasmic/neuropil with a few intranuclear locations, and found primarily in gray matter (Gutekunst et al. 1999) . The largest number of inclusions identified in patient tissue in those studies was observed in the deeper cortical layers, with many fewer and much smaller inclusions found in the striatum.
10.4 Cell Autonomous and Non-cell Autonomous Toxicity in Huntington’s Disease
The most prominent area of neurodegeneration in HD is the striatum. The dysfunction of the MSNs in this region and their ultimate degeneration is at the core of the motor abnormalities that exist in this disease. The striatum is the central input area of the basal ganglia receiving excitatory glutamatergic input from both the cortex and thalamus and dopaminergic input from the substantia nigra (Graybiel 1990; Wilson et al. 1990; Bolam et al. 2000) . Although the mHTT protein is expressed throughout the nervous system, much of the focus of HD research has been in the striatum. However, a series of studies in mice demonstrate the importance of other cell types in HD and their effect on neuropathological and behavioral manifestations of disease phenotypes in mice. There is clear evidence for non-cell autonomous mechanisms of toxicity in HD. In a mouse model with inducible expression of a mHTT-exon1 fragment (the Rosa/HD mouse), induction of expression throughout the brain (using Nestin-Cre) in neurons and glia, results in neuropathological abnormalities including gliosis and neurodegenerative changes in cortex and striatum. However, when the expression of mHTT expression was restricted to the cortex (using Emx1-Cre), or the striatum alone (using Dlx5/6-Cre) there were no significant behavioral or neuropathological changes at the ages examined (Gu et al. 2005, 2007) . In another model conditionally expressing a different mHTT fragment in MSNs under the control of the Darpp32 promoter (DE5 mice), there is late onset motor abnormalities but no evidence of neurodegenerative changes (Brown et al. 2008) , whereas mice expressing this fragment throughout the brain showed extensive neuropathological changes (Yu et al. 2003) .
10.5 Huntingtin Expression in Astrocytes
10.5.1 Expression in Human Astrocytes
The HTT protein is found throughout the nervous system. The majority of studies of pathogenesis in HD has centered on its dysfunction in neurons. However, RNA in situ hybridization identified positive signal in astrocytes from normal brain tissue (Landwehrmeyer et al. 1995) . Furthermore, brain tissue from HD patients that was stained with antibodies to HTT, and glial fibrillary acidic protein (GFAP) revealed the presence of HTT in astrocytes (Singhrao et al. 1998) although to a lesser degree than what is seen in neurons. Astrocytes in various brain regions including the striatum (caudate nucleus and putamen) as well as white matter from HD patients also contained mHTT positive aggregates (Singhrao et al. 1998; Shin et al. 2005; Bradford et al. 2009; Faideau et al. 2010) .
The Htt protein is also found in astrocytes from mice (Bradford et al. 2009; Lee et al. 2013) . The normal function of this protein in astrocytes remains to be completely elucidated. However, in mouse Hdh (encoding endogenous mouse huntingtin) knock-out neural stem cells treated using a neuronal differentiation protocol, there was a significant increase in the number of GFAP positive cells and a decrease in the number of microtubule associated protein 2 positive neurons when compared to control cultures. This finding suggests that wildtype Htt is involved in controlling the differentiation of neuronal and glial cells and that production of neurons from neural stem cells requires a normal level of wildtype Htt (Conforti et al. 2013) . This data is in line with previous studies showing that wildtype Htt plays a role in central nervous system development and neuronal survival (Reiner et al. 2003; Lo Sardo et al. 2012) . The wildtype Htt expressed in these stem cells could be acting in an instructive or repressive role, to promote neuronal fate and/or repress the glial fate. However, the exact role of wildtype Htt in astrocytes in the nervous system will need to be further assessed in conditional knock-in mouse models, where one can specifically reduce the expression of endogenous Htt only in astrocytes.
10.5.2 Expression in Mouse Astrocytes
Mutant HTT positive aggregates are found in astrocytes in the brains of HD mouse models. These mice contain aggregates in the white matter as well as the gray matter (Reddy et al. 1998; Yu et al. 2003; Shin et al. 2005) . Like the aggregation found in neurons in these mice, aggregation in the astrocytes also appears to be progressive, with the number of mHTT aggregates increasing as the animal ages. The aggregates are found in the striatum and cortex of these mice, but also in the corpus callosum (Shin et al. 2005; Bradford et al. 2010) . In addition, in a mouse model expressing a fragment of mHTT only in astrocytes, driven by the human GFAP promoter, there are aggregates in the astrocytes in the cortex, striatum, brainstem and spinal cord (Bradford et al. 2009) .
10.6 Astrogliosis in Huntington’s Disease
10.6.1 Humans
One prominent feature found upon neuropathological examination of HD patient tissue is the presence of astrogliosis (Vonsattel et al. 1985; Faideau et al. 2010) (Fig. 10.2). In neurodegeneration , it is generally believed that astrogliosis is a response to dysfunction or death of neurons. In HD patient brains, there is a significant increase in the number of reactive astrocytes as disease grade (neuropathological severity) increases (0–4). Astrogliosis was assessed in striatal tissue from all disease grades using GFAP immunohistochemistry. These studies reveal that GFAP immunoreactivity is present throughout the striatum in all disease grades. Furthermore, the GFAP level seems to increase as a larger number of astrocytes are expressing GFAP as disease grade increases (Faideau et al. 2010) . The pattern of astrogliosis in the striatal tissue from these patients seems to follow the pattern of neurodegeneration , where it is first seen in the dorsal striatum and then in the ventral striatum. Since the dorsal striatal astrogliosis is so early in the disease process (Grade 0), at least as far as obvious neuropathological changes are seen, this suggests that there is likely a cell-autonomous change in the astrocyte. The increase in GFAP staining intensity exists with characteristic reactive astrocytic phenotypes including hypertrophic cell bodies. As disease progresses the reactive phenotype becomes more severe with hypertrophic and overlapping protrusions from the astrocytes (Fig. 10.2; Faideau et al. 2010).
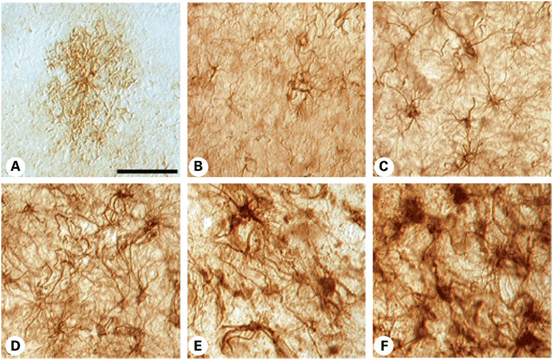
Fig. 10.2
GFAP immunohistochemistry in 50 µm tissue sections from the caudate nucleus in non-neurological control (a), and in increasingly severe HD specimens, Grades 0–4, Grade 0 (b), Grade 1 (c), Grade 2 (d), Grade 3 (e) and Grade 4 (f) HD subjects. Normal astrocytes present as faintly GFAP-stained cells with a short lace-like branching pattern distributed symmetrically around the cell soma. With increasing disease progression, there was greater GFAP immunoreactivity, twisting and thickened arbors, with larger somal size. The degree of astrogliosis became so great as to mask their individual appearance. The magnification bar in (a) represents 100 µm and is the same in all photomicrographs. (Reproduced with permission from Faideau et al. (2010); Oxford University Press)
10.6.2 Mouse Models
The reactive astrocyte phenotype has been observed in many of the mouse models expressing mHTT. Neuropathologically, these models display varying degrees of pathological changes, with regionally atrophy, cellular atrophy, dark neuron degenerative changes, and mHTT aggregation. There are multiple mouse models that express some form of the mutant protein, either a full-length or truncated protein by either a knock-in or transgenic approach. Many of these models also display some degree of astrogliosis. In the RosaHD/Nestin-Cre model, expressing mHTT throughout the nervous system in both neurons and glia , there is significant astrogliosis in the cortex and striatum (Gu et al. 2005). Studies of astrogliosis in models where the mHTT protein is expressed in the cortex with RosaHD/Emx1-Cre and striatum with RosaHD/Dlx5/6-Cre revealed no significant astrogliosis when mHTT was restricted to neurons (Gu et al. 2005, 2007) . The data from these models suggested that cell-cell interactions were important for the development of the reactive astrocyte phenotype.
In the mouse model HTT171-82Q, where mHTT expression was targeted to astrocytes in the striatum through lentiviral expression, there was an increase in GFAP staining and astrocytes exhibiting a reactive phenotype. This phenotype increased in severity as the animal aged, with an increase astrocyte soma size (Faideau et al. 2010) . This indicates a cell-autonomous affect of the mHTT protein within astrocytes, as this reactive phenotype is not due to the expression of mHTT in neurons. Thus reinforcing the idea that reactive gliosis is not merely a consequence or response to sick or degenerating neurons in neurodegenerative diseases . In the knock-in mouse model, Hdh CAG 150, there is extensive astrogliosis in the striatum (Lin et al. 2001) . Together, these mice demonstrate that the reactive astrocyte phenotype can be elicited from expression of mHTT specifically in the astrocytes or observed when mHTT expression is also found in neurons, therefore the mHTT protein is able to elicit both cell-autonomous and non-cell autonomous phenotypes.
10.7 Excitotoxicity in Huntington’s Disease
There are many possible mechanisms that may contribute to toxicity in HD; one of these is excitotoxicity . Excitotoxicity leading to neuronal dysfunction and death is caused by excessive activation of glutamate-gated N-methyl D-aspartate receptors (NMDARs) due to increased exposure to glutamate. This leads to Ca2+ overload and mitochondria energy failure (Coyle and Puttfarcken 1993) . This mechanism had been hypothesized for HD many decades ago. In HD this mechanism has primarily focused on the cortico-striatal synapse, with the pre and post-synaptic neuron receiving the most attention. The MSNs in the striatum receive glutamatergic excitatory input from both the cortex and thalamus (Fonnum et al. 1981a, b) . The excitotoxicity hypothesis of HD pathogenesis is supported by the existence of these extensive inputs and the presence of high densities of glutamatergic receptors in striatal neurons (Albin et al. 1990; Beal 1994; Landwehrmeyer et al. 1995) . Many of the initial studies in HD used excitotoxins to mimic HD pathology. One of the first rodent models of HD used injections of the excitotoxin kainic acid into the striatum to selectively destroy MSNs (Coyle and Schwarcz 1976; McGeer and McGeer 1976) . Quinolinic acid, a selective NMDAR agonist was also used to replicate the features of HD including selective degeneration and morphological changes in MSNs including loss of dendritic spines in rodents and non-human primates (Sanberg et al. 1989; Beal et al. 1991; Ferrante et al. 1993; Zeron et al. 2002) . This selective NMDAR agonist produced specific toxicities for striatal MSNs, without causing degeneration of striatal interneurons, further reinforcing the idea that excitotoxicity caused by activation of NMDARs as an important mechanism in HD .
The excitotoxicity hypothesis is further supported by data from multiple HD mouse models. There is increased response of MSNs to NMDAR activation. When quinolinic acid was injected into the YAC72 and YAC128 mice, there is a significant difference in lesion size as compared to wildtype mice, although as disease progressed in these models this phenotype did not persist in older mice (Zeron et al. 2002; Graham et al. 2009) . Furthermore, there is increased glutamate level in the striatum of YAC128 mice upon cortical stimulation (Joshi et al. 2009) , although other studies suggest no such change in young YAC128 mice not yet displaying behavioral or neuropathological features of HD (Milnerwood and Raymond 2007; Cummings et al. 2010) . These data support an altered NMDAR function early in the course of disease in these mouse models, and that altered striatal NMDAR signaling likely contributes to the deficits seen in HD .
10.8 EAAT2 Expression and Glutamate Level
10.8.1 Humans
The glutamate transporter is critical for regulating glutamate levels at the synapse. The uptake of glutamate and its conversion into glutamine reduces the level of glutamate in the synaptic space. In support of the excitotoxicity hypothesis, HD brains show a decrease in the level of the excitatory amino acid transporter 2 (EAAT2; human)/Glutamate transporter 1 (GLT-1; rodent) (Cross et al. 1986; Arzberger et al. 1997) . In situ hybridization studies on HD brain tissue reveals a decrease in EAAT2 mRNA labeling that correlated with disease severity (Arzberger et al. 1997) . In the tissue, a decrease in the number of cells expressing EAAT2 mRNA can be seen in the remaining tissue of both the caudate and putamen, although the putamen seemed to have the greater decrease. In addition immunohistochemistry performed on grade 0 to grade 4 tissues with an EAAT2/GLT-1 antibody revealed a grade dependent decrease in protein levels (Faideau et al. 2010). This data reveals that there is a loss of EAAT2 early in the disease process, which can implicate the transporter as a primary component in the initiation of disease. To properly maintain synaptic function and glutamate neurotransmission, there must be coordinated activity of the pre- and post-synaptic cells, but also the astrocyte. The role of this transporter in astrocytes is to remove glutamate from the synaptic cleft after it is released (Danbolt 2001; Maragakis and Rothstein 2001) . With excitotoxicity as one of the proposed toxic mechanisms in HD, due to increased levels of glutamate in striatal tissue that is hypothesized to come from the cortico-striatal inputs, the ability to efficiently uptake glutamate from the synaptic space is of vital importance.
10.8.2 Mouse Models
Based on the decrease in the level of the astrocyte specific glutamate transporter EAAT2/GLT-1, this has been a major focus of studies aimed at understanding astroyctic contribution to HD. Much of this work has been performed in multiple mouse models expressing various forms of the mHTT protein. There is a reduction in the level of Glt-1 mRNA in the R6/2 mice, which express an exon1 fragment with an expanded CAG repeat, when compared to littermates (Behrens et al. 2002; Shin et al. 2005) . The decrease in Glt-1 mRNA levels seem progressive as a decrease can be seen in R6/2 animals in 6 week old animals, in both cortex and striatum and further declines at 12 weeks (Behrens et al. 2002) . In addition to mRNA levels in the R6/2 mice, there is a significant reduction in the protein level of GLT-1 at 11–12 weeks of age (Behrens et al. 2002) , although there is a likely a decrease in protein levels early as well, it does not reach statistical significance (Shin et al. 2005) . Glutamate uptake is decreased in the striatum of the R6/2 transgenic model (Lievens et al. 2001; Behrens et al. 2002; Shin et al. 2005; Estrada-Sanchez et al. 2009) . Another mouse model, the Hdh CAG 150 knock-in mouse did not show a significant difference in the Glt-1 mRNA at 9 months of age although there was a slight reduction in Glt-1 mRNA levels (Shin et al. 2005) . This result is likely given the slower progression in this full-length mHTT mouse model that exhibits no obvious neuropathological changes at this age (Lin et al. 2001) .
Mice were generated using the human GFAP promoter to specifically express a mHTT exon 1 fragment carrying 160Q repeat in astrocytes (GFAP-HD) (Bradford et al. 2009). There is a significant decrease in the level of GLT-1 protein in the brains and cultured astrocytes from these mice. As a consequence of the decrease in the levels of Glt-1, there is also decreased glutamate uptake in the GFAP-HD mice (Bradford et al. 2009). In mice that expressed a mHTT fragment containing 82 CAG repeats (viral HTT-82Q) specifically in astrocytes using lentiviral vectors there was a decrease in the level of Glt-1 mRNA in the striatum of these mice 12 weeks after injection (Faideau et al. 2010). The level of the Glt-1 protein was assessed by immunohistochemistry in the mHTT positive astrocytes showed a significant and progressive decrease in the striatum. Interestingly, there was also a decrease in GLAST, but this was not significant until late in the disease process in this animal. Glutamate transport was also decreased in the astrocyte HTT171-82Q expressing mice and no decrease in glutamate transport or Glt-1 levels was observed with neuronal expression of the HTT171-82Q (Faideau et al. 2010). Perhaps the most interesting observation from these mice was the decrease in the levels of two neuronal proteins DARPP-32 and GluN2B subunits of NMDARs, thus indicating that the presence of mHTT in astrocytes is likely sufficient to alter neuronal activity and function. The exact mechanism whereby the mHTT in the astrocytes caused the decrease in the levels of these proteins is unclear, but implicates the inability of GLT-1 to properly buffer extracellular glutamate as a possible mechanism for decreased expression of DARPP-32 and GluN2B in neurons.
The mechanism for decreases in GLT-1 levels in the mouse models include a change in Sp1-dependent transcription of Glt-1 and palmitoylation of GLT-1. In mHTT expressing astrocytes from GFAP-HD transgenic mice, there is a reduction of the transcription factor Sp1 occupancy at the Glt-1 promoter as compared to littermate controls (Bradford et al. 2009). This reduction in this model is likely due to increased association of mHTT with Sp1 as shown by more mHTT precipitating with Sp1 than with HTT with a polyQ repeat in the normal range (Bradford et al. 2009). The YAC128 model expresses full-length human mHTT and recapitulates aspects of HD neuropathology and behavior that becomes progressively worse as the animal ages (Hodgson et al. 1999; Slow et al. 2003) . There does not appear to be a decrease in the levels of GLT–1 protein in the brain of these mice even as disease progresses. Interestingly, glutamate uptake was decreased in the striatum of these mice as early as 3 months of age, and at 12 months is also seen in the cortex (Huang et al. 2010) . EAAT2 was identified in a proteomics study of palmitoylation (Kang et al. 2008) , which involves the thioesterification of palmitic acid to cysteine residues and functions in tethering proteins to membranes or sorting them to lipid microdomains. Palmitoylation was reduced on GLT-1 in the brains of YAC128 mice (Huang et al. 2010) . The decrease in palmitoylation of GLT-1 was found to impair glutamate uptake, without affecting is localization to the membrane. Thus, exactly how palmitoylation affects the function of this receptor is still up for debate; nonetheless, these data provide two mechanisms for decreased GLT-1 levels and activity in HD mouse models.
With the alterations in GLT-1, studies have been performed to determine if increasing the expression of GLT-1 would alleviate phenotypes caused by the presence of mHTT. When Glt-1 was overexpressed by using a lentiviral vector in astrocytes also expressing HTT-171-82Q, the level of GLT-1 increased and the reactive astrocyte previously seen in those mice significantly decreased (Faideau et al. 2010). Furthermore, the use of ceftriaxone, a β-lactam antibiotic in R6/2 mice raised Glt-1 levels and reversed the glutamate uptake deficit in these mice. Ceftriaxone as improved some of the motor deficits found in the R6/2 mice. However, use of ceftriaxone must be approached with caution as it has been found to affect long term-depression in the hippocampus (Omrani et al. 2009) and impairs prepulse inhibition (Bellesi et al. 2009) . Nonetheless, while the appropriate approach to take has yet to be determined, increasing the levels of GLT-1 in HD may have beneficial affects on glutamate uptake deficit and motor impairment.
10.9 Glutamate Release from Mutant HTT Expressing Astrocytes
Much of the study in HD on astrocytes centers around the decrease in EAAT2/Glt-1 levels and the ability of the astrocyte to take up excess glutamate from the synaptic cleft and how that may contribute to excitotoxicity . However, there has been a lack of appreciation for the ability of the astrocyte to release glutamate and whether that ability is changed in HD. The astrocyte is the only cell in the brain that can synthesize glutamate de novo (Hertz et al. 1999) . They are capable of releasing glutamate through various mechanisms, including but not limited to Ca2+ dependent vesicular exocytosis (Parpura and Zorec 2010) . Glutamate released from astrocytes has been shown to act on extrasynaptic NMDARs to modulate neuronal excitability and synaptic transmission (Araque et al. 1999; Fellin et al. 2004) . Based on this ability of astrocytes to modulate neuronal excitability, it is possible that a change in this function due to the presence of mHTT in astrocytes could contribute to changes in the activity at the most critical excitatory synapses in the brains of HD patients.
To date only one study has been performed to determine whether mHTT expression in astrocytes had any affect on the levels of Ca2+-dependent glutamate release (Lee et al. 2013) . This study used cultured solitary astrocytes from the full-length human mHTT expressing BACHD mouse. This study employed a previously validated system where astrocytes were first grown in flasks culture and purified solitary astrocytes were then mechanically stimulated resulting in glutamate release (Hua et al. 2004) . Mechanical stimulation of astrocytes allows one to assess the exocytotic release of glutamate (Montana et al. 2004) . Mechanical stimulation of full-length mHTT containing cortical astrocytes from BACHD mice resulted in an increase in the level of glutamate released into the extracellular space near the solitary astrocytes as compared to wildtype astrocytes (Lee et al. 2013) . Astrocytes exhibiting Ca2+-dependent exocytotic release of glutamate do so by increased cytosolic Ca2+ responses which are usually in proportion to the level of glutamate released from these cells (Parpura and Haydon 2000) . While these BACHD cells responded to mechanical stimulation with a rise in cytosolic Ca2+ levels, there was not a significant difference between wildtype and full-length mHTT expressing cortical astrocytes in cytosolic Ca2+levels upon mechanical stimulation.
Multiple mechanisms could account for the change in levels of glutamate released from astrocytes. These include the trafficking of glutamate containing vesicles and glutamate synthesizing machinery within these cells. However, analysis of glutamate containing vesicles revealed no change in mHTT expressing astrocytes when compared to wildtype astrocyte, suggesting other mechanisms are involved to cause the increase in levels of glutamate released from these astrocytes.
Related posts:
 Experimental Treatment of Acquired and Inherited Neuropathies
Possible Therapeutic Targets in Microglia
General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
Enteric Glial Cells: Implications in Gut Pathology
Astroglial Connexins as Elements of Sleep-Wake Cycle Regulation and Dysfunction
Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Experimental Treatment of Acquired and Inherited Neuropathies
Possible Therapeutic Targets in Microglia
General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
Enteric Glial Cells: Implications in Gut Pathology
Astroglial Connexins as Elements of Sleep-Wake Cycle Regulation and Dysfunction
Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


