Fig. 14.1
Contributions of genetic and environmental factors to alterations in the CNS
Animal Models of ASD
BTBR Mouse Model
Animal models of human conditions allow researchers to directly test hypotheses generated from clinical observations. Development of effective animal models requires extensive research regarding the biological and behavioral pathologies of the condition. The BTBR mouse is recognized to be a valid genetic model of ASD. It is characterized by reductions in sociability, impaired social communication, and increased repetitive/compulsive behaviors that are the core features of ASD. Impaired sociability is observed through reduced social approach and social motivation during the three-chamber social approach task. BTBR mice also spend significantly less time engaging in social interactions, such as sniffing. Social communication deficits are observed through altered ultrasonic vocalizations (USV), both in frequency and quality. As olfactory cues are important in mouse communication, it is noted that the BTBR mouse also has fewer scent marking behaviors than the more typically developed C57BL/6J mouse model. The third core feature of ASD, repetitive behavior, restricted interests, and insistence on sameness, is also observed in the BTBR mouse model. BTBR mice spend excessive time engaging in self-grooming behaviors, and the behaviors themselves are altered and do not occur in the typical behavioral sequences. Perseverative behaviors are represented by an increased frequency of compulsive marble burying and persistent errors in the modified T-maze. These behaviors result in cognitive deficits, including impairments in reversal learning and reduced behavioral and cognitive flexibility. In addition to representing the core impairments of ASD the BTBR genetic mouse model also has a number of immune abnormalities, similar to the profile seen in children with ASD. This includes elevated expression of proinflammatory cytokines IL-1β, IL-6, and IL-12 in brain tissue, in addition to increased microglia activation, and increased numbers of microglia (Careaga et al. 2015). The expression of IL-33 in BTBR brain tissue has also been correlated with increased impairment in behavior (Heo et al. 2011). BTBR mice show skewing in macrophage and microglia subtype, with M1 skewing and increased production of IL-12 and less production of anti-inflammatory IL-10 (Onore et al. 2013).
The immune system abnormalities in this genetic model of ASD demonstrate a potential role for epigenetic changes as causative factors of ASD behaviors. Interestingly, when BTBR mice are exposed to a maternal immune activation (MIA) insult, there are more persistent immune alterations and more severe behavioral impairments. This is potentially due to a two-hit gene x environment interaction. Crosses between different strains of mice also demonstrate the role that maternal environmental factors and immune insults play in ASD etiology. For example, when the BTBR mouse is crossed with the healthy C57BL/6J mouse, both its immune and behavioral phenotypes are improved. Additionally, if BTBR embryos are placed in C57BL/6J dams, there are also significant improvements in both social and repetitive behaviors. On the other hand, when the C57BL/6J mouse is given an MIA insult, such as exposure to poly(I:C) its offspring resemble BTBR mice in both their immunological alterations and behavioral impairments (Careaga et al. 2015).
MIA Mouse Model
The MIA mouse is the only environmental model of ASD that has all three core impairments. The MIA insult varies, and can be exposure to IL-6, poly(I:C), LPS, or influenza, all with similar results in the immunological and behavioral effects on the offspring. Offspring of dams immune-activated with poly(I:C) have both the behavioral and neuropathological symptoms of human ASD in addition to altered immune profiles. Poly(I:C) MIA offspring have deficits in prepulse inhibition, a feature common in individuals with ASD, in addition to increased stereotypic behavior and decreased social preference. MIA offspring do not appear to have global changes in the levels of activated or total brain microglia/macrophages. Rather, dysregulated immune function in MIA offspring is characterized by systemic deficits in CD4+ TCRβ+ Foxp3+ CD25+ T regulatory cells, increased production of IL-6 and IL-17 by CD4+ T cells, and elevated levels of peripheral Gr-1+ cells. Regulatory T cells are suppressors of the innate and adaptive immune response, and deficits in these cells could result in the proinflammatory phenotype observed in both animal models and humans with ASD. If poly(I:C) MIA offspring are irradiated and transplanted with donor bone marrow from healthy saline exposed mice, their repetitive behavior deficits are normalized, and they have significantly less anxiety-like behavior. Social preference is not improved by the healthy bone marrow transplant. Interestingly, if irradiated poly(I:C) MIA offspring are transplanted with poly(I:C) affected bone marrow their repetitive and anxiety-like behaviors also normalize. Both types of transplants appear to immunologically normalize MIA offspring, suggesting that the peripheral environment of the MIA offspring is critical. These findings imply that some ASD-related behavioral abnormalities may be corrected by repairing immune function. However, the authors caution that transplant procedures may have their own ameliorative effects outside of the type of bone marrow used. In particular, irradiation could alter metabolic function, gastrointestinal microbial composition, and oxidative stress, all of which could directly influence behavior (Hsiao et al. 2012). Further detail about the MIA mouse model is provided below, in the Maternal Immune Activation section of this chapter.
MIA Non-human Primate Model
In addition to mouse models, non-human primate models are also used to study the longitudinal immunological and behavioral effects of MIA. Non-human primate models help to bridge the gap between the clinical population and rodent models due to their similarities with humans both genetically and behaviorally. Rodent models also have limitations in understanding the effects of MIA on fetal brain development, as the human third trimester occurs during the early postnatal period in rodents. Additionally, there are challenges in relating rodent brains and behavior to human brains and behavior. Recent MIA models have focused on challenges during the first and second trimester, which are known gestational windows of vulnerability for fetal development. Pregnant rhesus monkeys injected with poly(I:C) during the end of the first or second trimester had a strong inflammatory response, including sickness behavior, increased temperature, and altered cytokine profiles. MIA offspring were subsequently observed for 24 months. Initial observations of these primates showed increased distress and self-soothing behaviors and increased maternal attachment. When compared to control animals, the MIA offspring had more motor stereotypic and self-directed behaviors in addition to aberrant vocal communication and social interactions that mirror the core symptoms of ASD. Just as with rodent models, non-human primate models also have limitations, including small sample sizes and delayed brain pathology studies that may not reflect the mechanistic neural basis for behavioral abnormalities (Bauman et al. 2014).
Hygiene Hypothesis and Its Relationship to ASD
The hygiene hypothesis suggests that stimulation of the immune system by healthy gut microbes is protective against the development of inflammatory diseases, and that a rise in hygiene in urban settings has been associated with less protective microbes in humans and an increase in autoimmune inflammatory disorders such as multiple sclerosis, inflammatory bowel disease (IBD), asthma, allergic rhinitis, and potentially ASD. One of the major differences between infectious diseases in the developing world versus industrialized nations is the prevalence of parasitic infections (Rook 2008). Studies from the developing world have demonstrated that parasitic infection is statistically significantly associated with reduced risk of skin sensitization to allergens (Feary et al. 2011). Studies examining allergy status before and after anti-helminthic treatment showed an overall increase in skin allergen sensitivity after de-worming (Jouvin and Kinet 2012). Epidemiological studies in humans vs. the industrialized world have shown that infection with helminth parasites is associated with a lower incidence of allergy/asthma and autoimmunity. The primary factor associated with allergic and autoimmune disease is apparently loss of species diversity, both parasitic and symbiotic, from the human biome. In those with a genetic predisposition and adequate environmental triggers, the absence of species from the human biome, many of which co-evolved with the human immune system, leads to hypersensitive states that could foster allergic and autoimmune disease. The high prevalence of autoimmunity and allergy in the industrialized world and the well-established effects of inflammation on cognitive development supports the hypothesis that gene by environment (both external environment and gut microbiome) effects could lie at the center of the pathophysiology of ASD (Rook et al. 2014; Parker and Ollerton 2013).
The Microbiome and ASD
Definition and Roles of the Microbiome
It is estimated that up to 100 trillion microbial cells are found in the human body and that they outnumber human body cells by an order of magnitude. These cells as a whole are known as the human microbiome and recent research has demonstrated that these microbial species and their interactions with the host (human body) play key roles in homeostasis and the growth and continued development and function of the nervous system. Advances in technology have led to rapid DNA sequencing techniques, known as metagenomics, and have allowed for classifications of new microbial species and definitions of phylogenetic relationships between species (Mulle et al. 2013). This advancement in the understanding of the microbiome’s role in brain health and disease has contributed to our knowledge of the microbiome–brain–gut axis (Wang and Kasper 2014). Communication between the nervous system and the gut occurs through various mechanisms: (1) neural, including parasympathetic innervation, through the vagus nerve, and sympathetic innervation (2) endocrine, including the hypothalamic–pituitary–adrenal (HPA) axis (3) hormonal (4) metabolic, via nutrients and (5) immune-mediated mechanisms. Dysfunction of this axis is implicated in stress-related disorders such as depression, anxiety, and irritable bowel syndrome in addition to neurodevelopmental disorders such as ASD. Thus strategies that modulate the microbiome may be a novel therapeutic approach to developing treatments for neurodevelopmental, CNS and immune-related disorders (Mulle et al. 2013; Borre et al. 2014).
The gut microbiome is composed of bacteria belonging to two major phyla, Firmicutes and Bacteriodetes, and three enterotypes comprised of one of three genera, Bacteroides, Prevotella, and Ruminococcus. These enterotypes are largely related to dietary intake, in addition to host genetics, age, and other environmental factors. The core microbiome in healthy adults is typically developed by age 3, and functions in nutrient intake, immunological development, and disease pathology. Microbial species can break down plant polysaccharides and carbohydrates, and are believed to have evolved to help the human body extract the maximal energy supply from food sources. Certain species have also been shown to interact with the host immune system to produce intestinal T-cells and correct the balance of Th1/Th2 cytokine response (Mulle et al. 2013).
The symbiotic relationship between the immune system and the gut microbiome begins early in life, and is influenced by many factors, including breastfeeding, diet, antibiotics, birth order, and method of birth (cesarean or natural). There is also evidence that some microbial species may be passed from mother to child while in utero, and that prenatal stressors may further affect the development of a healthy microbiome (Borre et al. 2014). For instance, rhesus monkeys exposed to perinatal stressors have lower amounts of bifidobacteria and lactobacilli in adulthood (Bailey et al. 2004). Additionally, germ-free mice are shown to have increased behavioral abnormalities, altered gene expression, and altered HPA axis function that can only be corrected if they are reconstituted with normal microbiota early in life (Sudo et al. 2004). Thus, early life events modulate the microbiota and impact the ability of the immune system to create a healthy microbiome which aids in CNS development.
Alterations in the ASD Microbiome
Individuals with ASD have a reduced richness and diversity and altered composition of gut microflora (Kang et al. 2013). The first hypothesis of an impaired microbiome being linked with ASD was in 1998 (Bolte 1998). It was initially hypothesized that clostridia toxin could adversely affect neurotransmitter function and result in the neurobehavioral changes seen in ASD. Since then studies have repeatedly found abnormal levels of Clostridia species in those with ASD. Those with ASD and bowel problems have been found to have significantly increased levels of clostridium histolytica when compared to healthy controls (Goyal and Miyan 2014; Finegold et al. 2002; Song et al. 2004; Martirosian et al. 2011). Although another study in regressive ASD showed the reverse (lower levels of Clostridia species in ASD compared to controls), they did find that these children had significantly higher levels of Disulfovibrio species, and that these levels positively correlated with severity of autism symptoms (Finegold et al. 2010; Finegold 2011a, b). There is also a noted lack of the prevotella enterotype, which is key in digesting plant polysaccharides and carbohydrate-rich foods. This altered composition of the microbiome has been observed in multiple studies of children with ASD that compare them to both healthy controls and sibling controls. The lack of this enterotype may be linked to the deficiencies in disaccharide metabolism that are observed in some individuals with ASD (Kang et al. 2013). Insufficiencies in the digestion and absorption of disaccharides and monosaccharides could alter the balance of growth substrates by creating an environment more suited to competitive bacteria phylotypes that grow on undigested and unabsorbed carbohydrates (Williams et al. 2011). These changes in the intestinal environment have also been linked to the abnormal Firmicutes to Bacteriodetes ratios found in biopsies of children with ASD when compared to those with IBD (Goyal and Miyan 2014). In contrast, Sutterella spp. has been found to be increased relative to controls, both in fecal samples and biopsies of the gastrointestinal (GI) tract (De Angelis 2013). Although Sutterella was believed to be more prominent in those with GI disturbances, including those with ASD and GI disturbances and those with IBD, these results were not replicated in more recent studies, and it appears that Sutterella species may be more common to the human gut than previously thought (Williams et al. 2012; Hansen et al. 2013; Mukhopadhya et al. 2011; Wang et al. 2013, 2014; Parracho et al. 2005).
An attempted meta-analysis of the above studies demonstrates that despite small sample sizes and varied methodologies there are significant differences in the prevalence of different GI microbial species in children with ASD compared to controls, but further work is needed to better understand these differences and how they relate to ASD symptomatology and etiology (Cao et al. 2013). Overall, most studies have demonstrated an altered microbiome in patients with ASD, with the largest issue being a reduction in diversity and richness of microbial species when compared to controls. Increased diversity of the microbiome may allow for better microbial integrity, and the ability to protect the host from environmental stresses and reduce incidence of GI disorders and infections. There is a high prevalence of GI distress, between 23 and 70 %, in those with ASD, including abdominal pain, bloating, diarrhea, constipation, and food selectivity (Cao et al. 2013). Further study is needed to determine if the prevalence of GI symptoms in children with ASD is higher than typically developing children, however, data shows that those with ASD have higher prevalence rates of constipation (33.9 vs. 17.6 %) and food selectivity (24.5 vs. 16.1 %) than controls (Ibrahim et al. 2009). There may be a significant correlation between an altered microbiome and the GI symptoms present in ASD, but further study is needed (Jyonouchi et al. 2005a, b). However, GI symptoms have been linked to ASD symptom severity. For example, constipation, the most common GI symptom in ASD, is significantly correlated with increased irritability and insomnia (Kang et al. 2013; Adams et al. 2011; Buie et al. 2010; Coury et al. 2012; Hsiao et al. 2013). Approximately 90 % of children with ASD experience food selectivity, a form of behavioral rigidity that results in eating a narrow variety of foods by type, texture, or presentation. Issues with feeding may begin early in development, as breastfeeding is less prevalent and occurs for a shorter duration in those with ASD. This could be due to issues in metabolizing lactate due to an altered microbiome, or to difficulties in the child’s social engagement with the feeding process (Mulle et al. 2013; Williams et al. 2011). These deviations could further perpetuate an altered microbial ecosystem in ASD that is not remedied as the child develops. As noted above, a healthy and balanced microbiome is needed in order to properly digest foods, of note, di- and poly-saccharides. It is therefore possible that deviations in the establishment and maintenance of a healthy microbiome could result in difficulties in digesting certain foods, such as carbohydrates, resulting in GI distress and pain and leading to rigidity in food selections that maintain the altered microbiome. The irritability, social withdrawal, and anxiety symptoms observed more frequently in those with ASD and GI distress could thus be remedied by interventions that restore microbial balance.
Evidence of Altered Cytokine and Chemokine Levels in ASD
Brief Review of Cytokines
Cytokines are important modulators in both the initiation (innate immune system) and maintenance (adaptive immune system) of immune responses that also help to facilitate the exchange of information between cells of the immune system and other tissues. Involved in both proinflammatory and anti-inflammatory pathways, the effect of these proteins can cross the blood–brain barrier via active transport mechanisms. These proteins that are important in cell signaling include chemokines, interferons, interleukins, lymphokines, and tumor necrosis factor. They carry out the effector functions of T cells by altering the behavior of the target cells. All effector T-cells produce cytokines of different types in different combinations. These small secreted proteins and related membrane-bound proteins act through cell-surface receptors and induce changes in gene expression within their target cell (Masi et al. 2014; Parham 2009). Antigen-presenting dendritic cells take up pathogens and antigens within infected tissue and carry them in the lymph to the secondary lymphoid tissues where they stimulate T-cell activation by prompting naïve T cells to differentiate into effector T cells: CD4 Th1 cells and Th2 cells. Proinflammatory CD4 Th1 cells leave the lymphoid tissues and enter the blood to seek out the site of infection and stimulate the production of more effector T cells. When Th1 cells reach the site of infection they activate tissue macrophages for phagocytosis of microorganisms. Anti-inflammatory CD4 Th2 cells remain in the secondary lymphoid tissues where they activate naïve B cells specific to the antigen of the T cell which differentiate into antibody-secreting plasma cells. Th1 and Th2 CD4 cells are distinguished by the sets of cytokines they make and the effects that these have on the immune response. Proinflammatory Th1 cells work mainly with macrophages in developing a cell-mediated immune response, and secrete cytokines IFN-γ, TNF-α, IL-2. The activated macrophages will then secrete cytokines IL-1, IL-6, IL-8 (CXCL8), IL-12, and more TNF-α. Anti-inflammatory Th2 cells work only with B-cells to develop an antibody-mediated immune response, and secrete IL-4, IL-5, IL-6, IL-10, IL-13, and TGF-β. Once an infection has been brought under control by Th1 cells, Th2 regulatory cells act as immunosuppressors by preventing the production of new effector T cells and suppressing the functions of existing effector T cells. Of note, IL-6 can act as both a proinflammatory and anti-inflammatory cytokine depending on its route of production. An additional subpopulation of CD4 T cells is Th17 cells, which produce IL-17 (Parham 2009). These cells appear to have a proinflammatory role and have been linked to the inflammation seen in MS and IBD (Fleming et al. 2011). The relationship between proinflammatory and anti-inflammatory cytokines keeps the immune system at a balance, which, as described below, is shown to be disrupted in ASD (Fig. 14.2).
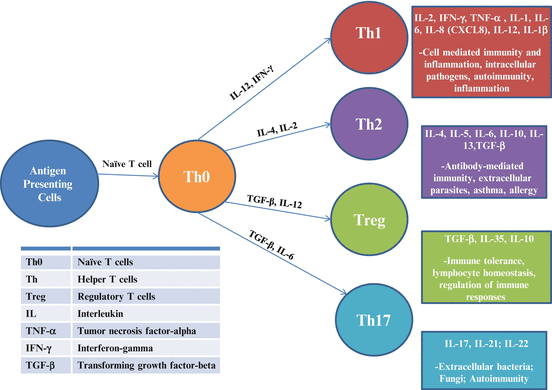
Fig. 14.2
Illustration of cytokine response. Note: Depending on context certain cytokines can act in Th1, Th2, or Treg roles
Cytokine Profiles of Individuals with ASD
Individuals with ASD have been observed to have altered cytokine profiles compared to healthy controls, and to be in a chronic state of cytokine induction. Many studies have shown those with ASD have a dampened Th2 anti-inflammatory cytokine response and an increased Th1 pro-inflammatory cytokine response, including an increased innate and adaptive immune response through the Th1 pathway, suggesting that localized brain inflammation and autoimmune dysfunction may be involved. More specifically, increases in IL-6, IL-8, and IL-23 have been observed, with an increased Th1 to Th2 ratio when compared to controls. These aberrations have been shown in amniotic fluid (elevated IL-4, IL-10, TNF-α, TGF-β), plasma, and peripheral blood mononuclear cells. Further studies have shown increased proinflammatory cytokines in the brain and cerebrospinal fluid (CSF) of ASD patients, including higher levels of proinflammatory IL-6, IL-8, IL-12 and interferon-gamma (IFN-γ) and tumor necrosis factor-α (TNF-α); in addition to elevated proinflammatory Th17 cytokines (Pardo et al. 2005; Abdallah et al. 2013a; Al-Ayadhi and Mostafa 2012; Masi et al. 2014; Croonenberghs et al. 2002a, b; Li et al. 2009; Ricci et al. 2013; Chez and Guido-Estrada 2010; Chez et al. 2007; Garbett et al. 2008; Goines and Ashwood 2013). This upregulation of cytokines has been linked to the activation of microglia and astroglia in ASD patients. Meta-analysis on studies that compare the plasma and serum concentrations of cytokines in unmedicated ASD and healthy participants found significantly altered concentrations of cytokines, further strengthening the hypothesis of an abnormal cytokine profile and immune system dysregulation in ASD that results in increased proinflammatory signals and CNS impairment. The largest effect sizes showed an elevation of proinflammatory IFN-γ and a reduction in anti-inflammatory TGF-β1, while increased levels of proinflammatory IL-1β, IL-6, eotaxin, IL-8, and MCP-1 were also significant. These cytokines have diverse and significant roles in the immune system. TNF-α is a proinflammatory cytokine involved in synapse maturation and stabilization, which at high levels has been shown to impair the development of the visual system by prematurely stabilizing developing synapses leading to increased local connectivity, and potentially epilepsy (Lee et al. 2010; Masi et al. 2014). IFN-γ is responsible for coordinating the transition from innate to adaptive immunity by amplifying immune system sensitivity and promoting macrophage activation. By skewing the immune response toward the Th1 phenotype IFN-γ inhibits the proliferation of Th2 cells and subsequently anti-inflammatory cytokines. TGF-β1 is an immunosuppressant required for maintaining homeostasis of the immune system in addition to promoting cell growth, proliferation, and differentiation (Masi et al. 2014). TGF-β is also a key regulator of neuronal C1q expression required to initiate microglia-mediated synaptic pruning and additionally modulates programmed cell death (Bialas and Stvens 2013). This is relevant to the hypotheses involving aberrant neuroconnectivity in ASD due to either excessive or insufficient synaptic pruning. IL-1β is known to affect the HPA axis and is involved in both acute and chronic inflammation. The proposed impaired chemokines in ASD, eotaxin, IL-8, and MCP-1 are involved in the recruiting neutrophils, eosinophils, monocytes, and T-cells to areas of tissue inflammation (Masi et al. 2014).
A Review of IL-6
IL-6 is an important neuroimmune factor that is shown to be involved in brain development and several neurological disorders. It can act to both promote neural growth and cause neuronal death, depending on its concentration, brain region, and the cell type affected. IL-6 facilitates the communication between the CNS and immune system, and has been linked to the impairment of neural cell adhesion and migration, synapse formation, and synaptic plasticity. Elevated levels have been linked to detrimental effects on synaptic plasticity in addition to physiological and pathological effects on learning and memory. Its role in promoting neuronal differentiation in the adult hippocampus may be compromised if it is chronically elevated, as seen with altered levels of synaptic proteins in the hippocampus during chronic IL-6 administration. Alterations in neural cell adhesion and migration may lead to an imbalance of inhibitory and excitatory synaptic transmissions, including elevated excitatory synapse formation and impaired inhibitory synapses, resulting in reduced paired-pulse inhibition and short-term synaptic plasticity. The impaired excitatory/inhibitory balance theory is one of the leading hypotheses in ASD etiology, and the role of IL-6 demonstrates that it may be linked to immune-inflammatory hypotheses that have spurred recent research. In fact, IL-6 is also proposed as a key factor in the MIA mechanistic pathway (Wei et al. 2013).
Neonatal Cytokine Levels in ASD
The abnormal levels of cytokines and chemokines in patients with ASD may indicate a pathological inflammatory disease process that could result in increased susceptibility to pathogens and allergens resulting in abnormalities in the immune system and CNS impairments. Additional research demonstrates that these abnormalities may begin early in life, with neonatal blood samples showing depressed immune cell activity, including decreased levels of IFN-γ, IL-2, IL-4, IL-6 and Regulated upon Activation Normal T-cell Expressed and Secreted (RANTES, or CCL5) and increased levels of IL-8 and soluble IL-6 alpha. Increased levels of these chemokines, MCP-1 and RANTES, were also observed in the plasma of ASD children. However, as seen with other etiological factors of ASD, results across studies are inconsistent. An additional study of neonates only found increased levels of MCP-1 and decreased levels of RANTES (CCL5) compared to healthy controls, and did not report changes in other cytokines or a hypoactive immune system. Nevertheless, the altered levels of these chemokines that are key to healthy neuronal migration could alter neurodevelopment. Studies that demonstrate links between MIA and developmental delays and ASD features in offspring support this hypothesis (Abdallah et al. 2012, 2013b; Zerbo et al. 2014).
Associations Between Cytokine Levels and Behaviors
CNS impairment may be indicated in ASD through the severity of behaviors and diagnostic features. Similar to evidence of the altered microbiome, an altered inflammatory response, observed as elevated cytokines, has also been linked to ASD symptoms. Worsening GI symptoms have been shown to aggravate behavioral symptoms. Those who have fluctuating behavioral symptoms following immune insults have additional innate immune abnormalities and significantly altered transcript profiles of peripheral blood monocytes (Jyonouchi et al. 2011). Constipation, the most prominent GI symptom in those with ASD, has also been associated with increased levels of blood myeloid dendritic cells (Breece et al. 2013). These cells act as messengers between the innate and adaptive immune system, and present antigen material to the T-cells of the immune system. Higher levels of these cells is also associated with increased repetitive behaviors in those with ASD, larger amygdala volumes, developmental regression, and a later onset of ASD symptoms. Typically developing children with altered dendritic cell frequencies are shown to have poorer adaptive behavior skills. Associations with the amygdala are relevant to ASD etiology, as it is implicated in alterations of core social abilities in ASD and the modulation of anxiety, in addition to having a role in modulating GI activity (Breece et al. 2013). Reduced adaptive behavior skills were also associated with lower plasma levels of TGF-β1, a crucial regulator of the immune system and mediator in the development of autoimmune and systemic inflammation. These children with ASD and low plasma levels of TGF-β1 were found to have worse behavioral scores on the Aberrant Behavior Checklist (ABC) including higher levels of irritability, social withdrawal, hyperactivity, and stereotypies. These results were seen in both children with early onset ASD and those who had regressed, but not for typically developing children or those with developmental disabilities (Ashwood et al. 2008). Other studies have also shown no differences in immune abnormalities between those with early onset ASD and those with developmental regression (Ashwood et al. 2011b). Both groups have a skewed Th1/Th2 response to stimulation with phytohemagglutin (PHA), including increased levels of Granulocyte-macrophage colony-stimulating factor (GM-CSF), TNF-α and Th2 cytokine IL-13 and reduced Th1 cytokine IL-12p40. Increases in Th2 cytokine production are associated with better behavior scores on the ABC (IL-13), improved adaptive behaviors on the Vineland Adaptive Behavior Scale (VABS), and better developmental and cognition scores on the Mullen Scales of Early Learning (MSEL) (IL-10 and IL-5). In contrast, increases in Th1 proinflammatory cytokines were associated with severity of ASD behaviors on gold standard measures of ASD, the Autism Diagnostic Interview-Revised (ADI-R), the Autism Diagnostic Observation Schedule (ADOS), and aberrant behaviors on the ABC. The improvement in behaviors and cognition observed to be associated with increased GM-CSF is significant, as it can cross the blood brain barrier and act as a neuronal growth factor and promote axonal regeneration. However, these receptors are also found on microglia, astrocytes, neurons, and oligodendrocytes, and blocking GM-CSF suppresses microglia activity and the neuroinflammation seen in disorders such as Alzheimer’s disease and multiple sclerosis. In contrast, other studies have showed higher cytokine profiles in those with ASD and regression compared to those with ASD without regression (Ashwood et al. 2011c). Both populations had higher levels than typically developing children, and higher levels of Th1 proinflammatory cytokines IL-1β, IL-6, IL-8, and IL-12p40 were associated with severity of core behaviors on the ADI-R and more severe aberrant behaviors on the ABC, in particular increased stereotypical behaviors. Levels of chemokines MCP-1, eotaxin, and RANTES observed to be increased in the plasma of ASD children compared to typically developing and developmentally disabled controls were also associated with severity of behaviors, cognition, and adaptive function observed on the MSEL, ABC, and VABS (Ashwood et al. 2011a).
Cytokine Levels in Siblings of Individuals with ASD
Of note, siblings of children with ASD have been shown to have immune profiles that are significantly similar to ASD profiles. This autism endophenotype in unaffected siblings was proven in a recent study that compared multiple immune parameters between the two groups. Both children with ASD and their siblings had increased levels of proinflammatory cytokine IL-6, and anti-inflammatory cytokine IL-10 (Saresella et al. 2009). This study and others (Manzardo et al. 2012; Napolioni et al. 2013) highlight that the immune dysregulation observed in ASD is also visible in siblings, indicating the need of further research into genetic and environmental insults that may affect both populations. It also further emphasizes that siblings should not be used as healthy comparators in studies of ASD immune dysfunction.
Summary
The altered cytokine profile, in addition to observations of altered T-cell activation, increased circulation of autoantibodies to brain tissue, increased monocyte cell activation, natural killer (NK) cell abnormalities, skewed immunoglobulin profiles, and alterations in complement components, suggest a definite underlying immune dysfunction in ASD (Hsiao 2013; Goyal and Miyan 2014). There has been significant variation in the results seen in these studies, which could be due to varied experimental designs, diagnostic criteria, ages of probands and control populations, use of non-clinically assessed siblings as controls, and small sample sizes. Nevertheless, a definite immune dysfunction is present, and ongoing inflammatory responses may be linked to disturbances in behavior and cognition in ASD.
The Role of Microglia in Inflammation and ASD
Definition of Microglia
The genesis, development, and functional remodeling of our highly complex central nervous system requires strict homeostatic control. This homeostasis is maintained by neuroglial cells, which include macroglia (astroglia, oligodendoglia, and NG2 cells) and microglia. Microglial cells originate from myeloid progenitors that enter the CNS during early embryonic development. They are the first and only glial cells populating the early embryonic brain and have multiple physiological functions that are important for the development and shaping of synaptic connectivity. Their functions include (1) early synaptogenesis during which microglia can provide growth factors and thrombospondins, (2) the elimination of redundant synapses, (3) the direct modulation of synaptic transmission through secretion of factors such as BDNF or TNF-α, (4) provision of trophic support, and (5) the regulation of neurogenesis. Microglial cell bodies are relatively stationary, but their processes continually probe tissue, so that complete coverage of the brain is completed every few hours as they search for signs of damaged tissue (Zeidan-Chulia et al. 2014). Additionally, expression of IL-6, IL-1β, and TNF-α, cytokines observed to be increased in ASD, is correlated with microglia activation (Tetreault et al. 2012; Frick et al. 2013). In sum, they are resident immune cells in the CNS that serve to detect damage, secrete cytokines, and control neuroinflammation, in addition to having a significant role in synaptic pruning during critical periods of development (Zhan et al. 2014; Pardo et al. 2005).
Microglia Pathology in ASD
Microglial pathology has been suggested to have a role in neurodevelopmental disorders such as ASD. Functional connectivity appears to be decreased across cortical regions and is linked to decreased expression of genes associated with synaptic transmission and abnormalities in synaptic scaffolding proteins. Multiple hypotheses about the etiology of ASD revolve around circuit-level deficits in the disorder, including the underconnectivity model, weak central coherence model, and the enhanced excitatory–inhibitory model. It is possible that microglial abnormalities in ASD lead to deficits in synaptic pruning during critical periods of development resulting in connectivity issues that lead to behavioral and cognitive deficits. Microglia’s role in synaptic elimination also provides a link between the ASD hypotheses involving the immune system’s role in etiology, and those revolving around synaptic connectivity (Zhan et al. 2014). Questions remain about whether activated microglia serves to protect or harm within patients with ASD. Typical neurons prevent phagocytosis by microglia by emitting the cytokine protein fractalkine (CX3CR1) (Tetreault et al. 2012; Zhan et al. 2014; Derecki et al. 2014). Mouse models that delete the microglial fractalkine receptor have more dendritic spines in CA1 neurons and reduced microglia-mediated synaptic pruning which results in abnormal brain development and immature connectivity. Additionally, disorders where microglial pruning is impaired, such as Nasu-Hakola disease, are characterized by deterioration of social behavior and dementia (Tetreault et al. 2012). This suggests that microglia typically have a neuroprotective role, and disruption can cause widespread neuronal damage that can result in neurobehavioral deficits.
Post-mortem studies of the brains of individuals with ASD demonstrate a marked increase in neuroglial responses, characterized by activation of the microglia and astroglia and increased production of both proinflammatory and anti-inflammatory cytokines. This occurs in the absence of lymphocyte infiltration and immunoglobulin deposition. Although microglial responses were diffusely distributed across the cortex and subcortical white matter, there is significant neuroglial activation in the cerebellum. This is of particular interest in ASD due to neuropathological studies that indicate abnormalities in both cortical organization and neuronal packing, and reduced cerebellar Purkinje cell numbers (Vargas et al. 2005). Microglial cells are also described to be of increased density in the dorsolateral prefrontal cortex of post-mortem ASD brain tissue (Morgan et al. 2010, 2012). Additionally, there are significant increases in average microglial somal volume in white matter, microglial density in gray matter, and trends of increased microglial somal volume in gray matter. Microglia also appear to be closer to neurons in the dorsolateral prefrontal cortex in ASD. These changes were present across age groups, including children under the age of 6, during periods of known brain overgrowth and macrocephaly in ASD. Soma enlargement characterizes a shift toward the amoeboid morphology of microglia, a sign of activation and differentiation towards a cytotoxic phenotype. ASD brain tissue also has increased microglial densities in the visual cortex and the fronto-insular cortex. Visual cortices play a role in the deficits in social cognition and social communication observed in the early years of ASD as reduced eye contact, deficits in visual tracking, and prolonged and perseverative fixation. The fronto-insular cortex is involved in social behaviors and is shown to have reduced activity in those with ASD (Tetreault et al. 2012).
In Vivo Microglia Activation Study in ASD
In the first in vivo study of microglia activation in ASD, both men with ASD and age and IQ matched controls received positron emission tomography (PET) using the microglia radiotracer [11C] (R)-PK11195. As observed in the post-mortem brain studies, there was an increased microglia activation across a wide range of brain areas, including the brainstem, anterior cingulate cortex, orbitalfrontal cortex, midfrontal cortex, superior temporal cortex, fusiform, parietal cortex, and corpus collosum. The most prominent increase in microglial activation was observed in the cerebellum. The pattern of distribution of [11C] (R)-PK11195 binding potential values was similar between the ASD and control subjects, which could indicate augmented but not altered microglial activation in ASD. Correlation studies between levels of microglial activation and severity of behaviors initially were not significant. However, subtyping the subjects by level of binding potential (high BP versus low BP) revealed more severe behaviors on the ADI-R and ADOS in the high BP group. The limitations of this study may have contributed to the minimal links between microglial activation levels and clinical features. As described by the authors, the group was higher functioning and did not include subjects with the most severe ASD behaviors, or those that had a history of regression or immunologic abnormalities. Additionally, the radiotracer used has a high incidence of non-specific binding (Suzuki et al. 2013). Previous studies also suggest that microglial alteration in ASD may be heterogeneous to the disorder, just as many of the clinical features are. Nevertheless, this initial in vivo study provides further evidence of immune alterations and increased microglial activation in those with ASD.
Microglia in Rett’s Syndrome, an X-Linked ASD
Interestingly, ground-breaking research in Rett’s syndrome, an X-linked ASD, further demonstrates the importance of microglial phagocytic activity and posits bone marrow transplantation as a possible therapeutic approach. Rett’s syndrome is characterized by the mutation of the MeCP2 gene, which is attributed to neuronal dysfunction, including altered glia. When the Rett’s syndrome mouse model (MeCP2-null) is irradiated and receives a bone marrow transplant from healthy wild-type mice, bone-marrow derived myeloid cells of microglial phenotype engraft brain parenchyma, resulting in the stoppage of disease development. However, when the MeCP2-null mouse does not receive cranial irradiation, the engraftment by healthy microglia does not occur, and the disease development continues. Additionally, when MeCP2 mice with newly engrafted microglia receive annexin V, a pharmacological agent known to block the phagocytic activity of microglia, the benefits of arresting disease development are diminished. Although future studies need to be conducted to better understand the roles of microglia and their phagocytic activities play in Rett’s syndrome, the amelioration of symptoms in this mouse model and the link between this disorder and ASD are significant to therapeutics developments for both disorders (Derecki et al. 2012).
Summary
In sum, microglia are necessary to modulate synaptic activities and plasticity and to aid in the structured remodeling of neuronal circuits. They may play different roles depending on the spatial and temporal context of the afflicting neurodevelopmental or neurodegenerative disease and they may be both a response to the underlying condition and a cause of further abnormalities and insult (Wu et al. 2013). It is clear that they are important in the pathology of ASD, but further research is needed to clarify their role at each developmental stage, and how this may result in aberrant neuronal connectivity and resulting behavioral abnormalities.
The MIA Hypothesis and ASD
Animal Models of MIA
MIA during pregnancy is an environmental risk factor for psychiatric illnesses, such as ASD, in offspring. The effect of MIA on offspring neurodevelopment and behavior has been demonstrated in multiple animal models using a variety of immune system activators, including the influenza virus, the viral mimetic polyinosinic-polycytidylic acid (polyI:C), IL-6, and the bacterial endotoxin lipopolysaccharide (LPS). Early prenatal stress (EPS) can also act as an immune system activator, and has been shown to produce changes in gene expression patterns, including the up-regulation of immune-related genes of proinflammatory cytokines and chemokines such as IL-6, IL-1β, MCP-1, MIP-1α, and CCL5 (RANTES) (Bronson and Bale 2014). The timing of the MIA is critical to the subsequent effect on the fetus, with early first trimester and second trimester insults causing the most significant insult (Garay et al. 2013). Alterations in brain cytokine levels are found throughout the lifespan of MIA offspring. In both the frontal cortex and cingulate cortex, proinflammatory and anti-inflammatory cytokines are elevated in the early postnatal period, lower than controls during early adolescence, and elevated again in the adult brain. The hippocampus also has alterations of cytokine levels at each age level, but these are distinct compared to other brain areas. There also appears to be a widespread decrease in cytokine levels during critical periods of synaptogenesis and plasticity in MIA offspring compared to controls. This is in contrast to the expected pro-inflammatory phenotype and may indicate an alternative pathway for cytokines to impact brain connectivity and subsequently behavior in ASD. Of note, brain cytokine levels are different than serum cytokine levels, indicating that the levels in brain tissue are not contaminated by serum. Additionally, MIA offspring have no changes in BBB permeability, immune cell infiltration, or microglial density (Garay et al. 2013; Patterson 2012; Ito et al. 2010; Shi et al. 2009; Arrode-Brusés and Brusés 2012).
Offspring of MIA mice exhibit the three core features of ASD, including deficits in communication, sociability, and the presence of repetitive and stereotyped behavior. These ASD features are demonstrated through alterations in their USV, including fewer harmonic and more complex and short syllables, fewer USVs in response to social encounters; reduced scent marking; and deficits in marble burying and self-grooming (Malkova et al. 2012). MIA offspring also have alterations in fear regulation, with marked deficits in the recall and extinction of context. This is an indication of behavioral inflexibility, or rigidity, and is a common feature of the ASD phenotype (Sangha et al. 2014). The MIA model is the only model of an environmental risk factor for ASD, where all three of the core features are present. MIA with either influenza, poly(I:C), or IL-6 causes gene expression changes in the fetal brain transcriptome, most notably in the upregulation of α, β, and γ crystalline gene families (Garbett et al. 2012). This upregulation is correlated with the weight of the placenta, which is an indicator of the severity of MIA, and may be an attempt of the developing brain to counteract the environmental stress of the insult. Crystallin is a protein and neurite promoting factor involved in neuronal differentiation and axonal growth that may also be involved in neuroprotection in response to immune activation. Brain tissue of ASD patients has been shown to have altered αβ-crystallin protein in the frontal cortex. Additionally, altered crystallin levels are implicated in neurodegenerative disorders, and αβ-crystallin knockout mice used to study Alzheimer’s disease or autoimmune encephalitis are shown to have worsening symptoms, further indicating the role of crystallin as a neuroprotector. Upregulated crystallin appears to be transient, and characteristic of a response to the MIA exposure, as adolescent mice do not have these higher levels. However, this neuroprotective response appears to have detrimental consequences, and could alter neurogenesis and differentiation in the embryonic brain, causing delays in development that result in changes to connectivity and neurochemistry (Garbett et al. 2012).
Gene expression patterns across MIA offspring also demonstrate the importance of IL-6 as a critical mediator of the behavioral and transcriptional changes in the offspring. As mentioned above, IL-6 is significant in ASD pathology as it is elevated in both the CNS and peripherally. Additionally, when injected with IL-6 prenatally, adult mice exhibit behavioral and cognitive deficits. IL-6 may act at different locations in the MIA model, most likely at one or more of three major sites of signaling: the maternal immune system; the maternal/fetal interface (the placenta); and the fetal brain (Hsiao and Patterson 2011). IL-6 genes are upregulated in response to MIA or EPS in the placenta and in the fetal brain, although this increase may occur acutely (Pratt et al. 2013; Bronson and Bale 2014). After MIA the placenta shows increases in both IL-6 mRNA and the maternally derived IL-6 protein (Hsiao and Patterson 2011). IL-6 could alter the vascular permeability of the placenta, and hence the transfer of nutrients, hormones, or other key molecules to the fetus. Additionally, IL-6 could degrade the maternal tolerance of the fetus by activating lymphocyte migration and cytotoxicity, and enhancing the production of maternal antibodies that cross into the fetal brain. Interestingly, IL-6 could result in changes to fetal programming. Typically there is an increase in TGF-β in pregnant women that is responsible for developing lymphocytes into T regulatory cells that inhibit maternal responses which could be harmful to the developing fetus. TGF-β in combination with IL-6 may result in these cells instead being differentiated into TH17 cells in the offspring. TH17 cells produce pro-inflammatory cytokines, such as IL-1β, IL-6, IL-12, and TNF-α, which could further contribute to the neuroinflammation observed in those with ASD (Mandal et al. 2011). High levels of maternal IL-6 are required to see the behavioral changes observed in MIA offspring, as when IL-6 is blocked and eliminated from the maternal immune response to infection or insult, both gene expression and behavioral changes are almost completely normalized (Smith et al. 2007). However, certain types of MIA insults may require IL-6 for the survival of the mother. If mice mothers injected with influenza are given an anti-IL6 injection, they become severely ill and often die or have a miscarriage. Additionally, IL-6 KO mice that are infected with influenza have the same results, with changes in body temperature, weight loss, and anorexia. Poly(I:C) MIA mice may one of the few that can tolerate the removal of IL-6 from the immune response (Smith et al. 2007). Contrary to the above, an additional study of IL-6 KO mice showed that offspring have increased levels of inflammatory cytokines and chemokines compared to offspring of wild-type saline injected mice, even without an inflammatory insult. This heightened level of inflammation in IL-6 KO offspring does not increase when mothers are injected with poly(I:C), suggesting that the already inflamed brain tissue is not further insulted as a result of MIA (Pratt et al. 2013). The heightened inflammatory state of offspring of IL-6 KO mice is accompanied by higher levels of choline acetyltransferase (ChAT) and decreased expression of glutamic acid decarboxylase (GAD)65, even without MIA. These results are also observed in MIA offspring and are accompanied by an increase in the number of cholinergic neurons in the basal forebrain, an area that is known to have enlarged neurons in those with ASD. Associations between these changes and heightened cytokine production have been made, but it is unclear if this is a result of microglial activation. Decreases in GAD65 expression with heightened ChAT activity could be representative of skewed differentiation of GABAergic interneurons and hence alterations in ASD connectivity and excitatory/inhibitory balance (Pratt et al. 2013). The role of IL-6 in these alterations could indicate it interferes with the normal function of microglia, or, alternatively, it should act as a shut-off switch to regulate the inflammatory environment. IL-6 is also involved in the pathophysiology of epilepsy and may increase seizure susceptibility in offspring following MIA. This is relevant to ASD due to the comorbidity of epilepsy and epileptogenic activity. Inflammatory factors are known to be involved in epilepsy. Brain inflammation facilitates seizures through both enhanced excitation and compromised inhibition. MIA is shown to increase hippocampal excitability, prolong the persistence of kindling states, and increase the speed of the progression of kindled seizures, in addition to impairing social interaction. Both IL-6 and IL1-β are required for this increased seizure propensity, and their activity creates lasting changes in the offspring neocortex and hippocampus. Blockades of either of these cytokines during pregnancy remove the increased propensity for seizures in MIA offspring (Pineda et al. 2013). The ability of IL-6 to cause neuronal survival or neuronal degeneration, balance neurogenesis and gliogenesis, and affect brain development, learning, and memory and the CNS response to injury, clearly indicate it has a complex role in fetal development that requires further investigation.
Related posts:
 Experimental Human Endotoxemia, Sickness Behavior, and Neuropsychiatric Diseases
Developmental Immune Activation Models with Relevance to Schizophrenia
Role of Autoimmunity and Infections in Tourette Syndrome
The Role of Inflammation in Alzheimer’s Disease
Experimental Human Endotoxemia, Sickness Behavior, and Neuropsychiatric Diseases
Developmental Immune Activation Models with Relevance to Schizophrenia
Role of Autoimmunity and Infections in Tourette Syndrome
The Role of Inflammation in Alzheimer’s Disease
 The Role of Infections and Autoimmune Diseases for Schizophrenia and Depression: Findings from Large-Scale Epidemiological Studies
The Role of Infections and Autoimmune Diseases for Schizophrenia and Depression: Findings from Large-Scale Epidemiological Studies
 Animal Models Based on Immune Challenge: The Link to Brain Changes and Schizophrenia
Animal Models Based on Immune Challenge: The Link to Brain Changes and Schizophrenia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


