Fig. 4.1
Cross section of a brain capillary, depicting the main components
An important component in the protection of the brain against toxic influences is the multispecific efflux pump P-glycoprotein. This pump, a 170 kD protein, located at the luminal side of the capillary endothelial cells, has a large capacity and is capable of extruding a wide array of structurally divergent substrates. The brain uptake of the majority of antidepressants and antipsychotics, as well as many other psychotropic drugs and endogenous compounds, is hampered by the activity of P-glycoprotein.
In the following paragraphs, the main components of the BBB will be discussed. Subsequently, relevant issues in reference to the function of P-glycoprotein and other efflux pumps in the blood–brain barrier related to several major psychiatric disorders (mood disorders, stress-related disorders and neurodegenerative disorders, such as dementia) are addressed, such as a possible role of P-glycoprotein as a susceptibility factor in depressive disorders. Furthermore, the current state of knowledge concerning the role of P-glycoprotein on pharmacokinetics of psychiatric drugs and the impact of modulation of P-glycoprotein on major psychiatric disorders will be discussed.
4.2 Components of the BBB
4.2.1 Endothelial Cells
Brain endothelial cells (EC) are unlike other endothelial cells in the body. Compared to the leaky capillaries formed by mesenteric endothelium, the brain endothelium is 50–100 times tighter, as indicated by the transendothelial electric resistance (TEER) (Pardridge 2005). They differ phenotypically by a lack of fenestrations and the presence of tight junctions. The rate of pinocytosis is minimal and free membrane diffusion applies mainly to small lipophilic molecules like ethanol or nicotine (Pardridge 2002b). The ECs are surrounded by a basal lamina, which further restricts the microvascular integrity (Hamann and Schimrigk 1995; Pardridge 2002b). ECs are rich in mitochondria, necessary for their high metabolic demands. Specific enzymes expressed by ECs (monoamine oxidases, epoxy hydrolase, endopeptidases, etc.) are important elements, constituting the so-called metabolic barrier, and participate in the regulation of brain penetration of drugs and their metabolism (Henry and Duling 1999; Leung et al. 2009; Pardridge 2002a). ECs have an extensive transport system on their surface, which is carrier mediated or receptor mediated.
4.2.2 Tight Junctions
Tight junctions are the closely associated areas of two cells whose membranes join together forming a virtually impermeable barrier to fluid. This is accomplished by their structure, as it is composed of a branching network of sealing strands, a complex of transmembrane (junctional adhesion molecule-1, occludin and claudins) and cytoplasmic proteins (Hawkins and Davis 2005; Jiang et al. 2008). Each strand acts independently from the others. The tight junctions prevent the passage of molecules and ions through the space between cells, the so-called paracellular flux. In order to pass the blood–brain barrier, molecules must enter the endothelial cells. Together with the endothelial cells, the tight junctions play the most substantial role in maintaining the BBB.
4.2.3 Astrocytes, Microglia and Pericytes
Astrocytes, microglia and pericytes surround the endothelial cells and form the ‘second line of defence’ in the BBB. Astrocytes are star-shaped glia cells that perform many functions, including the biochemical support of endothelial cells. Their end-feet, encircling the endothelial cells, aid in the maintenance of the blood–brain barrier, as they regulate the homeostasis of brain water and electrolytes (Bernacki et al. 2008; Choudhuri and Klaassen 2006; Hawkins and Davis 2005). In addition, they interconnect endothelial cells with surrounding neurons. Astrocytes secrete proteins that can have an opposite effect on neurons and endothelial cells. For example, thrombospondin, an astrocyte-derived protein, stimulates neurogenesis on the one hand while it counteracts the effects of angiogenesis (Choi and Kim 2008). Current in vitro cellular models for the study of BBB function often incorporate astrocytes with endothelial cells. Studies have shown that when astrocytes are removed from an in vitro BBB model, an increased permeability is observed (Hamm et al. 2004). Many transporters (see below) are under the control of astrocytes (Omidi et al. 2008). In pathological conditions, astrocytes mediate the immune response; they synthesise proinflammatory cytokines and chemotactic factors, which eventually trigger the breakdown of the BBB (Jiang et al. 2008). The pericyte, another foremost cell type in the CNS, intimately embraces the brain endothelial capillary. Less is known about their role at the BBB, although they seem to stabilise the formation of capillary-like structures when they are added to a coculture of astrocytes and endothelial cells (Ramsauer et al. 2002).
Microglia are omnipresent throughout the brain parenchyma. They are the immune effectors in the CNS, as they can release a large number of immunoregulatory, inflammatory and cytotoxic mediators (Dheen et al. 2007). They surround the brain capillaries and stay in a resting state until they are activated, which can be induced by a variety of stimuli (Dijkstra et al. 2004; Guillemin and Brew 2004). Figure 4.1 shows the main components of the BBB.
4.2.4 Neurons
Neurons lie in close proximity of the brain capillaries and their needs, including protection and the unrestricted provision of nutrients, are optimally met. Neuronal tissue needs an abundant supply of oxygen. At rest, 80–92 % of its ATP comes from oxidative metabolism of glucose (Mintun et al. 2001). Given the dynamic nature of neural activity and the considerable metabolic needs of nervous tissue, the microcirculation of the brain must be highly responsive to the tissue it supplies. Iadecola found that blood flow was increased in response to local neuronal activation and proposed a role for nitric oxide as a messenger between blood and neuronal tissue (Iadecola 1993). This ‘metabolic coupling’ of regional brain activity to blood flow is the basis of functional neuroimaging. However, data from positron emission tomography (PET) and functional MRI studies indicate that during short-term functional activation, cerebral blood flow and cerebral metabolism (oxygen consumption) are not directly linked (Fox et al. 1988; Mintun et al. 2001).
4.3 Transport Across the BBB
There are two tightly controlled ways of transport for molecules and cells across the BBB (see Fig. 4.2). The paracellular route, or junctional route, is restricted by the interendothelial tight junctions. Tight junctions not only restrict paracellular flux but also maintain the polarity of enzymes and receptors on luminal (blood) and abluminal (brain) membrane domains (Matter and Balda 2003; Wolburg and Lippoldt 2002). Due to the presence of tight junctions, only lipid-soluble substances or those transported through an active mechanism can cross the BBB (Bernacki et al. 2008).
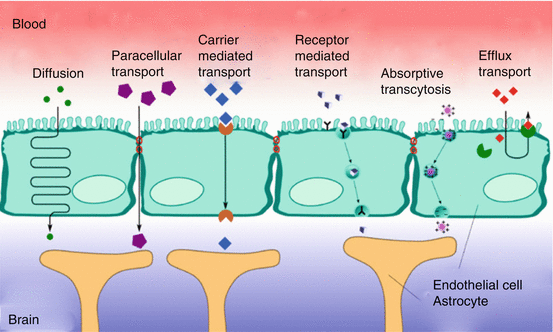
Fig. 4.2
Transport mechanisms at the blood–brain barrier (Adapted from http://upload.wikimedia.org/wikipedia/commons/2/24/Blood-brain_barrier_transport.png (no copyright))
The transendothelial route further restricts the passage of molecules by three distinct transport systems. Small lipid-soluble molecules can penetrate the brain EC through diffusion, unless their molecular weight exceeds 400–600 Da (Pardridge 2006). All other transport to the brain is via endogenous catalysed transport systems on the capillary membrane. As we will describe below, three main transport systems function at the BBB: (1) carrier-mediated transport, which relies on a molecular carrier present on both the luminal side and the abluminal side of the BBB; (2) receptor-mediated transport for endogenous large-molecule peptides such as insulin (Duffy and Pardridge 1987) or transferrin (Jefferies et al. 1984); and (3) active efflux transporters such as P-glycoprotein and many other active efflux transport systems within the BBB (see Fig. 4.2).
4.3.1 Carrier-Mediated Transport
Carrier-mediated transport is a highly selective form of transport for small molecules such as ions, glucose and amino acids. Examples of carrier-mediated transport systems include the GLUT1 glucose transporter, the OAT (organic anion transporter) and organic anion-transporting polypeptide family (OATP), the monocarboxylate transport family and the LAT1 (large neutral amino-acid transporter) (Tsuji 2005). This form of BBB transport is a saturable process which can be unidirectional or bidirectional. Carrier-mediated channels can be gated and can function ion and energy independent, like the LAT1 (Smith and Stoll 1998). The membrane spanning pores are highly stereospecific. The substrate forms a complex with the carrier in order to be translocated to the opposite side of the membrane. L-DOPA is such a drug that utilises the LAT1 to enter the brain. Once transported through the BBB, L-DOPA is reformulated to dopamine (Kanai and Endou 2003). Another example is valproic acid, which is delivered to the brain via a medium-chain fatty transporter (Adkison and Shen 1996). Uptake of valproic acid was reduced in the presence of medium-chain fatty acids, but not short-chain fatty acids, indicating that valproic acid is taken up by a transport system for medium-chain fatty acids. The monocarboxylate transport family also appears to be involved in the transport of valproic acid, possibly as efflux transporter (Adkison and Shen 1996; Tsuji 2005).
4.3.2 Receptor-Mediated Transport
Another main transport mechanism at the BBB is receptor-mediated transport, which involves a vesicular trafficking system of the endothelial cells (Fig. 4.2). The influx of several brain nutrients like leptin (Ziylan et al. 2009) and insulin (Matter and Balda 2003) occurs by this form of transport. Circulating molecules are bound to a specific receptor at the plasma membrane, to form a receptor-ligand complex. When the ligand is bound to the receptor, the process of invagination is initiated. Dependent on subsequent intracellular processes, the vesicles are either sent to the basolateral side of the cell, where they are released, or the complex is dissociated from the ligand in the cell. A similar transport system is called ‘absorptive-mediated transcytosis’ which does not require specific binding to a receptor. Instead, binding is nonspecific, for example, to negative charges on the plasma membrane. The receptor-mediated transcytosis offers a promise as drug vector for drug delivery into the brain (Jones and Shusta 2007).
4.3.3 Efflux Transport
Uncharged, small lipophilic compounds enter the brain much better than other molecules, because they can diffuse passively across the endothelial cells. However, an ATP-driven efflux system exists at the luminal wall of the capillary wall, which can extrude a variety of structurally diverse drugs, drug conjugates and metabolites and other compounds from the cell. These efflux proteins involved belong to the superfamily of ABC (ATP-binding cassette) transporters, an extensive and functionally highly diverse family of membrane transporters. Export of these compounds occurs in an active, ATP-dependent manner and can take place against a considerable concentration gradient. The human ABC transporter is classified into eight subfamilies (ABCA–ABCH) (Dean 2009). P-glycoprotein (P-gp, ABCB1), multidrug resistance protein ABCC subfamily (formerly denoted as MRP), ABCC4 and ABCC5 (and possibly ABCC1 and ABCC2; see below) (Dean 2009; Yu et al. 2007; Zhang et al. 2004; Zhou et al. 2008) and the breast cancer-related protein (BCRP, ABCG2) have been localised in the apical membrane of the brain endothelial cell and have a role in efflux at the BBB. Of these, P-gp is the best-studied transport protein. Its most striking property is transport of a wide range of structurally different substrates, including many CNS drugs. As a consequence, the net penetration of substrate drugs and other substrate compounds from the blood into the brain tissue can be dramatically decreased. Various members of the ABCC family show considerable differences in their tissue distribution, substrate specificity and proposed physiological function. These proteins play a role in drug disposition and excretion and thus are implicated in drug toxicity and drug interactions. ABCC primarily transports anionic compounds, such as glutathione S-conjugates and oxidised glutathione (Jedlitschky et al. 1994). Furthermore, ABCC transport appears to be dependent upon intracellular glutathione (Renes et al. 1999).
4.4 Localisation and Function of P-gp
Of all efflux pumps discovered at the BBB so far, P-gp is the best described. It has been shown that P-gp exerts an important influence on the penetration of psychoactive drugs at the BBB (Linnet and Ejsing 2008; Liu et al. 2008). P-gp is found at the luminal side of the endothelial cells, where it extrudes hydrophobic compounds from the cell. It was discovered in 1976 in drug-resistant ovary cells from Chinese hamsters (Juliano and Ling 1976). Human P-gp is encoded by the multidrug resistance gene (MDR1), which is now denoted as ABCB1 (ATP-binding cassette gene B1). P-gp contains 1,280 amino acids and has a molecular weight of 174 kD (Sharom 2006).
Cerebral P-gp is present not only at the BBB but also at the blood-CSF barrier (Rao et al. 1999). Apart from the CNS, P-gp is present in kidneys, gut and other organs that have an epithelial lining. Interestingly, while in normal conditions P-gp is not found in neurons, neuronal P-gp expression was reported in pathological conditions, such as refractory epilepsy, cortical dysplasia and glioneuronal tumours (Lazarowski et al. 2004; Volk et al. 2004; Aronica et al. 2003). The physiological function of ABCB1 has not been unambiguously identified yet (Banerjee and Bhat 2007; Sharom 2006), but it involves the protection of the brain from compounds that have gained access to the circulation. The fact that it is being expressed in damaged neuronal tissue, as well as in malignancies, affirm such a function (Lazarowski et al. 2004; Volk et al. 2004). It needs to be considered that xenobiotic or drug concentrations in the brain might also be affected by intestinal P-gp limiting oral bioavailability.
Its imposing ability to transport hundreds of structurally divergent drugs, natural products and peptides renders this protein a fascinating molecule. P-gp is composed of two transmembrane-bound domains, each consisting of six transmembrane helices and two nucleotide-binding sites, which hydrolyse ATP, enabling substrate transport (see Fig. 4.3) (Aller et al. 2009). P-gp substrates are known to partition into the lipid bilayers and accumulate to high concentrations. In the inner leaflet of the lipid bilayer, the compounds can then get in contact with the drug-binding pocket of the transporter (Aller et al. 2009; Banerjee and Bhat 2007). Initiated by ATP binding, a conformational change of the transporter molecule then brings the drug-binding site to the outer leaflet or extracellular surface, thereby promoting unidirectional transport to the extracellular space.
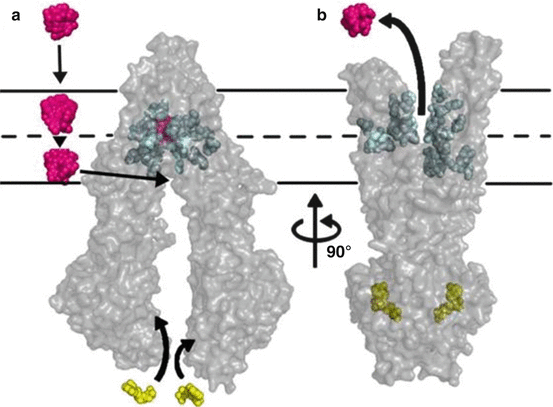
Fig. 4.3
Model of substrate transport by P-gp. (a) Substrate (magenta) partitions into the bilayer from outside of the cell to the inner leaflet and enters the internal drug-binding pocket through an open portal. The residues in the drug-binding pocket (cyan spheres) interact with a substrate in the inward-facing conformation. (b) ATP (yellow) binds to the nucleotide-binding domains, causing a large conformational change presenting the substrate and drug-binding site(s) to the extracellular space. In this model of P-gp, exit of the substrate to the inner leaflet is sterically occluded, providing unidirectional transport to the outside (Reprinted with permission from Aller et al. (2009))
4.5 In Vivo Imaging of P-gp at the BBB
In the last decade considerable effort has been put into gaining further insight into the role of P-gp under pathophysiological conditions. Different substrates of P-gp have been radiolabeled and imaged using single-photon emission computed tomography (SPECT) and positron emission tomography (PET) (Kannan et al. 2009).
A P-gp tracer that meets the criteria of a useful ligand is a selective substrate for P-gp, produces a good signal after P-gp blockade and generates few radiolabeled metabolites that contribute to the PET signal. The principle of PET imaging of P-gp activity amounts to measure the difference in cerebral uptake of a radiolabeled P-gp substrate (i.e. radioligand) under two different conditions, for example, major depressive disorder versus healthy control, or before and after drug inhibition. The function of P-gp can be quantified by calculating the distribution volume (V T) of the ligand, which inversely reflects P-gp function (Lubberink et al. 2007). V T is an estimate of the brain tissue-blood partition coefficient of activity of the radiotracer at equilibrium and is defined as the influx rate constant k 1 over the efflux rate constant k 2.
Several radiolabeled P-gp substrates are available for PET, such as [11C]-verapamil and [11C]-carvedilol, [11C]-loperamide and [11C]-desmethyl-loperamide (Elsinga et al. 2005; Hendrikse et al. 1998; Seneca et al. 2009). Of these, the in vivo studies with [11C]-verapamil outnumber all other ligands and it is the only tracer used in clinical studies (Elsinga et al. 2005; Lubberink et al. 2007; Sasongko et al. 2005). The (R)-enantiomer of [11C]-verapamil can be considered superior to the racemic mixture. Although the feasibility of in vivo measurement of P-gp function by [11C]-verapamil PET (VPM-PET) has been confirmed by several research groups (Lee et al. 2006; Lubberink et al. 2007; Sasongko et al. 2005), it is not an ideal tracer, because its brain uptake is low. The production of several metabolites of the parent compound [11C]-verapamil does not appear to be a problem, since the main metabolite is also a P-gp substrate (Lubberink et al. 2007). Several strategies have been launched in search of an optimal tracer that is capable of detecting subtle changes in function (in particular increased P-gp activity). The development of a radiotracer that is not a substrate but works as a pure inhibitor could bypass this problem, but so far the results of this strategy are ambiguous, possibly due to the fact that these inhibitors have affinity as a substrate too (Syvanen and Hammarlund-Udenaes 2010). Several new imaging strategies may still hold the unfulfilled promise of a method to study drug interactions with P-gp, for example, radiolabeling of a new drug candidate after administration of an inhibitor or a double PET scan to evaluate the effect of a P-gp modulator. Another strategy that could work is the co-administration of a P-gp inhibitor that can increase the baseline signal (Wagner et al. 2009). This new strategy may enable accurate estimation of dose–response relation of a given drug or facilitate prediction of drug interactions at the BBB (Bauer et al. 2012; Kreisl et al. 2010).
4.6 Relevance of P-gp in Relation to Antidepressants, Mood Stabilisers and Antipsychotics
Different in vitro approaches and in vivo models (ABCB1ab (−/−) knockout mice) have been used to assess the impact of P-gp on pharmacokinetics of psychiatric drugs (O’Brien et al. 2004). In vitro data have indicated that most antidepressants and antipsychotics have affinity for P-g (Maines et al. 2005; Störmer et al. 2001; Szabo et al. 1999; Weber et al. 2005; Weiss et al. 2003). Many of the in vitro data are contrary, which is inherent to the different methods and materials used.
Most research focusing on the inhibitory effects of antidepressants and antipsychotics has been done with various cell lines expressing (recombinant) human P-gp. As a measure of the P-gp-inhibitory potency of the drug, a prototypic P-gp substrate, such as calcein-AM or rhodamine 123, was often used. The concentration needed to displace 50 % of the prototypic compound (called IC50 or EC50) was used as measure of the inhibitory effect on P-gp activity.
For most compounds, the ability to inhibit P-gp has been compared to a typical comparator, such as verapamil or cyclosporine A. It must be considered that these inhibitors significantly differ in their potency to affect P-gp transport function (Achira et al. 1999). For example, haloperidol appeared to be a weak inhibitor compared to PSC833 (Wang et al. 2006), whereas it proved to be a much stronger inhibitor in comparison to verapamil and ivermectin (El Ela et al. 2004).
Some antidepressants have only shown inhibition without demonstrated substrate affinity (i.e. desipramine, imipramine, reboxetine) (Szabo et al. 1999; Weber et al. 2005). Besides, the translation of in vitro results to the human case could be complicated by differences in substrate specificity for P-gp across species (Baltes et al. 2007). A more reliable method of studying affinity for P-gp is based on studies in knockout mice, which lack the ABCB1ab genes encoding P-gp. Affinity for P-gp has been demonstrated in knockout mice for most of the common antidepressants (Doran et al. 2005; Ejsing et al. 2006; Störmer et al. 2001; Uhr et al. 2000, 2003; Uhr and Grauer 2003) and antipsychotics (Doran et al. 2005; Ejsing et al. 2005; Kirschbaum et al. 2008; Schinkel et al. 1996; Wang et al. 2003, 2004, 2009).
Despite the differences in the experimental set-up that partly explain the controversies regarding the interaction between P-gp and antidepressants and antipsychotics, it is feasible to say that the majority of the antidepressants and antipsychotics have shown (mostly weak) affinity as a P-gp substrate and that most have a weak inhibitory effect on P-gp in vitro. Contradictory to the results of most serotonergic antidepressants, St. John’s wort, a phytopharmaceutical with antidepressant effect, has shown in vitro inductory effect (Ott et al. 2010). Some authors have argued that the inhibitory effect of antidepressants on P-gp is clinically irrelevant (Uhr and Grauer 2003; Weber et al. 2005; Weiss et al. 2003), since the drug concentrations used in most studies necessary for P-gp inhibition were far above the therapeutic dose range. On the other hand, others have suggested that some antidepressants may be strong inhibitors in vivo (Peer et al. 2004; Störmer et al. 2001; Wang et al. 2008). For antipsychotics comparable differences between the individual drugs have been described.
The impact of cerebral P-gp in man for the bioavailability of psychoactive drugs cannot be satisfactorily deducted from the in vitro data. It needs to be considered that antidepressants and antipsychotics can generally access the brain and therefore cannot be high-affinity substrates of P-gp-like compounds that do not exhibit any relevant brain penetration based on its interaction with P-gp. Moreover, CNS-active drugs are in general characterised by a fairly high lipophilicity. Recent data demonstrated that it can be more difficult to detect an interaction with P-gp when drugs pass membranes efficaciously by rapid diffusion due to their lipophilic characteristics (Luna-Tortos et al. 2008).
Most of the antidepressants and antipsychotics, as well as several mood stabilisers, demonstrated a significantly greater, albeit small, brain/plasma ratio in ABCB1a knockout mice compared to wild-type mice (mean = 1.8 for antidepressants, 4.9 for antipsychotics). The value of the studies using the knockout mouse model is limited by the fact that a one-dose model is mostly used, representing only the acute modulatory effect on P-gp. There are only two reports on long-term treatment on P-gp activity in vivo (Grauer and Uhr 2004; Kirschbaum et al. 2010). For amitriptyline, it was demonstrated that the brain uptake in WT and KO mice was equal for the parent compound (amitriptyline), whereas a much higher cerebral concentration was measured for its metabolites in the knockout mice. It was suggested that chronic administration of amitriptyline induces upregulation of the P-gp pump, thus inhibiting its own access to the brain. In an elaborative study, they refined their results and demonstrated that 4 h after a single dose of amitriptyline, the initial differences (present at 1 h post injection) in cerebral uptake had disappeared, in contrast to the metabolites, pleading against an upregulatory mechanism (Uhr et al. 2007). Differences in cerebral uptake between the parent compound (amitriptyline) and its metabolites can also be explained by a differential affinity of P-gp for the respective molecules. Miura et al. demonstrated that small molecular changes can turn a strong P-gp substrate into a weak one (Miura et al. 2007). Another notion is that the effect of a non-substrate on P-gp activity can apparently vary over time (Störmer et al. 2001).
In spite of the findings of the preclinical work on the role of P-gp in the uptake of drugs used for the major psychiatric disorders, there are arguments pleading against a significant role for P-gp. For example, antipsychotic drugs like risperidone and haloperidol may significantly differ in their interaction with P-gp but have a comparable clinical efficacy.
Because direct evidence for a major role of P-gp in pharmacokinetics has been lacking, CNS side effects of drugs with potential P-gp inhibiting effects may erroneously have been attributed to other causes. Cytochrome P450 3A4, a major drug metabolising enzyme, shows a striking overlap in substrate specificity with P-gp (Fromm 2004; Zhou et al. 2008). Several CNS effects have been ascribed to interactions between drugs concurrently inhibiting Cyp3A4, without notice of a potential concomitant P-gp inhibiting effect of these drugs in vitro (Gelisse et al. 2007; Jover et al. 2002; Sheehan et al. 2006).
Cortisol is a substrate of P-gp as well. Its uptake into the brain is thwarted by P-gp (Karssen et al. 2001; Ueda et al. 1992). It has been well established that depression coincides with dysregulation of the HPA axis, characterised by negative feedback inhibition and elevated cortisol levels (Holsboer 2000). As some of the in vitro data have suggested that P-gp is inhibited by the action of antidepressants (Pariante et al. 2001), it has been hypothesised that antidepressants exert their antidepressant effect partly by the inhibition of P-gp, leading to an intracerebral cortisol increase and normalisation of the HPA axis. However, antidepressants appear to have an insignificant effect on plasma cortisol levels (Weber et al. 2006). Besides, cortisol entry to the brain is not regulated by P-gp only, as Mason et al. found in mice (Mason et al. 2010). In conclusion, data supporting the hypothesis that P-gp inhibition plays a role in normalisation of a hyperactive HPA axis are lacking.
Mood stabilisers belong to different drug classes. Lithium is the drug of first choice in the treatment of bipolar disorders. Although lithium itself is not a substrate of P-gp, lithium shares many properties with magnesium (Mg2+), such as a synergistic effect on the Na/K ATP-ase (Murck 2002). Mg2+ appears to play a key role in P-gp-mediated efflux through ATP hydrolysis (Singh et al. 2009). It may thus be conceived that lithium has a similar impact on the function of P-gp. Other drugs registered as mood stabilisers include the antiepileptic drugs carbamazepine, valproic acid and lamotrigine. These agents appear to have no, or at most a weak, affinity for P-gp. Reports on the effect of different antiepileptic drugs on P-gp expression in brain capillary cell lines have been equivocal (Ambroziak et al. 2010; Lombardo et al. 2008; Yang et al. 2008).
4.7 Modulation of P-gp at the BBB by Stress (In Vitro Studies)
An understanding of the physiological regulation of P-gp is key to therapeutic strategies in the treatment of psychiatric disorders, since most of the drugs used for depression and psychosis seem to be P-gp substrates. Stress is the hallmark of many psychiatric diseases, including depression and psychosis, and it appears that all sorts of stress responses evoked by a variety of environmental stimuli, such as cytotoxic agents, heat shock, irradiation, genotoxic stress and inflammation are able to influence either the expression or the activity of P-gp (Sukhai and Piquette-Miller 2000). P-gp is regulated at various levels of expression including DNA, mRNA and protein.
There is increasing evidence for the role of cytokines in the pathogenesis of depression (Schiepers et al. 2005). Proinflammatory cytokines are produced by different immune cells upon presentation of an antigen, and their secretion is believed to be the prime event in the subsequent neurophysiological responses taking place during immune stimulation. Several animal models are applied (Sukhai and Piquette-Miller 2000; McRae et al. 2003), and although results of the in vivo studies are somewhat conflicting, most demonstrated that P-gp expression and activity can be involved in different ways during an inflammatory episode, the degree and direction of the change in P-gp activity depending on the model and the inflammatory mediator used (McRae et al. 2003; Roberts and Goralski 2008).
Several in vitro studies have tried to extricate the signalling pathways of the involved cytokines and other chemotactic compounds leading to a functional change of P-gp at the BBB. Hartz and colleagues define a pathway through which P-gp is acutely modulated. They describe a sequence of events, starting with TNFα (tumour necrosis factor), releasing endothelin-1, which in turn activates nitric oxide synthase and then protein C kinase, ultimately reducing P-gp transport (Hartz et al. 2004; Hartz et al. 2006). The same group showed that this regulation is biphasic: after an initial rapid decrease of P-gp activity following exposure to TNF and endotheline-1, an increase in transport activity was found at 6 h post exposure. Similar data are presented by others. For example, the results by Tan and colleagues showing an increase in P-gp expression following BBB breakdown by activated T-cells suggest both a role in immune cell-mediated cytotoxicity and a counterregulatory role in promoting cell survival and maintaining BBB integrity. They hypothesise that the latter role may be relevant in later stages of the inflammation (Tan et al. 2002). In vivo studies focusing on such a biphasic response in cytokine regulation and in P-gp activity in depressive or psychotic disorders are warranted and can be best worked out in animal models.
4.8 Depressive Disorder, Stress-Related Disorders and P-gp
A few studies have focused on BBB characteristics in relation to psychological stress. Rats subjected to perinatal stress had an increased permeability as measured by the uptake of Evans blue (Gomez-Gonzalez and Escobar 2009). Acute stress (e.g. immobilisation stress or forced swim) increases BBB permeability likewise (Sinton et al. 2000; Chandler et al. 2002; Esposito et al. 2001). In respect to the effect of chronic stress on BBB function, studies are lacking, but in major depressive disorder (MDD), BBB dysfunction has been suggested as a potential mechanism (Gudmundsson et al. 2007).
If P-gp is a susceptibility factor in stress-related disorders, animal models could be used to demonstrate their modulatory role in stress. To date, evidence is sparse. In one study, describing the anxiolytic-like effect of tariquidar (a P-gp inhibitor) in mice sensitised by a mild stressor, it was suggested that P-gp inhibition led to an increase of corticosteroids, which, in turn, would enhance the negative feedback control of the HPA axis (Thoeringer et al. 2009).
In two PET studies with [11C]-verapamil, an increased function of P-gp at the BBB in temporal and frontal areas was found both in a group of medicated patients with MDD and schizophrenia (de Klerk et al. 2010b; de Klerk et al. 2009). Caution in the interpretation of the results of both studies is justified, since the findings were complicated by the use of either antidepressants or antipsychotics. The increase in P-gp function might be a result of the disorder itself or a result of antidepressant treatment. A sequel to these studies was set up to disentangle the possible contribution of antidepressant therapy and MDD. Rats were subjected either to chronic stress or to a continuously administered antidepressant. Using [11C]-verapamil PET as a measure of P-gp activity, an indication was found for decreased P-gp activity in the stressed rats, whereas venlafaxine appeared to have an opposite effect on P-gp activity (de Klerk et al. 2010a). A confirmation study correlating the in vivo results from both studies to P-gp expression at protein and mRNA level is under way. Although the in vivo imaging study may provide an indication for a susceptibility role of P-glycoprotein in depressive disorders, in the light of the limited evidence so far, it is impossible to interpret the findings without great restrictions.
4.9 The Role of Ageing and Neurodegeneration in P-gp Functionality
Although the status of both MDD and schizophrenia as neurodegenerative disorders is tenuous, both disorders have neurodegenerative features. In depression, it has been hypothesised that neuroinflammation ultimately leads to neurodegeneration (Maes et al. 2009). The coincidence of structural brain changes and decreased regional blood flow has been well established in MDD as well as in schizophrenia. Loss of the integrity of the blood–brain barrier is a cardinal phenomenon in many neurodegenerative disorders including Alzheimer dementia. In the PET studies on Parkinson’s disease a role for P-gp in neurodegeneration has been suggested, but this is based on small differences in tracer uptake between patients and control group (Bartels et al. 2008b).
The decreased P-gp function in old age, which has been indicated by [11C]-verapamil PET studies, will certainly accentuate the problems encountered in pharmacotherapy in the elderly (Bartels et al. 2008a; Bauer et al. 2009). The decline in activity might be linked to the pathology in late-life depression such as the cognitive changes. A major role of P-gp is however refuted by the fact that the incidence of MDD decreases with age (Kessler et al. 2003).
4.10 Polymorphisms of P-gp and Their Effect on Function
Since the first report on a polymorphism in the ABCB1 gene, the quest for more variations in the DNA sequence explaining phenotypical differences has not stopped. Substantial progress has been made in identifying single nucleotide polymorphisms (SNPs) in the entire ABCB1 gene. Reports in the literature have particularly focused on C3435T (noncoding exon 26), after an initial report on altered duodenal P-gp expression and functionality associated with the TT variant (Hoffmeyer et al. 2000), but later on the SNPs G2677T/A and C1236T have generated interest as well.
Studies within the field of psychiatry have particularly focused on treatment response to antidepressants or antipsychotics. Other studies refer to side effects, and two studies pertain to the occurrence of ABCB1 polymorphisms in mood disorders and schizophrenia. In two studies, the diagnosis of depression was found to be associated with a haplotype of 13 ABCB1 polymorphisms (Dong et al. 2009). In the other study, mood disorders were associated with the haplotype of 129-2677-3435 (T-A-C) and with a lower frequency of ABCB1 alleles at −1517, −41 and −129 and a higher frequency of 2677A (Qian et al. 2006). In this study no association was found between ABCB1 and schizophrenia.
Most studies on the genetic effects of the SNPs C3435T, G2677T/A and C1236T or ABCB1 haplotypes showed little or no effect on treatment response (Table 4.1). A strong effect of a single transporter gene on a phenotypic response in complex disorders like MDD and schizophrenia is not amenable. A few reports on antipsychotic- or antidepressant-related side effects associated with polymorphisms have been published, and a few groups have communicated on the effect of ABCB1 polymorphisms and plasma concentrations of antipsychotics or antidepressants (Table 4.2). For risperidone, displaying high substrate affinity for P-gp, only modest genetic effects were reported in side effects (weight gain) and in treatment response (Kuzman et al. 2008; Xing et al. 2006).
Table 4.1a
Treatment response to antidepressants and antipsychotics related to ABCB1 polymorphisms
Drug | DSMa diagnosis | ABCB1 variant | rs-idb | n cases c | Outcome measured,e,f,g,h,i | References | p-valuej |
|---|---|---|---|---|---|---|---|
Amitriptyline | G2677T/A | rs2032583 | 50 | % change in HAMD response after 3 weeks | Laika et al. (2006) | n.s. | |
Amitriptyline, venlafaxine or citalopram | MDD | Intron 4 | rs2235015 | 133 | % change in HAMD-21 score after 4 weeks | Uhr et al. (2008) | 0.0024 |
(C/T) intron 22 | rs2032583 | 132 | 0.00007 | ||||
Desipramine | MDD | UTR | rs17064 | 272 | % change in HAMD-21 score after 8 weeks | Dong et al. (2009) | n.s. |
Haplotype block 1 | Remission versus non-remission after 8 weeks | 0.02 | |||||
Paroxetine | MDD | (C/T) intron 22 | rs2032583 | 124 | % change in HAMD-21 score after 8 weeks | Sarginson et al. (2010) | 0.024 |
Intron 4 | rs2235015 | n.s. | |||||
(A/G) intron 21 | rs2235040 | 0.028 | |||||
MDD | G2677T/A | rs2032583 | 68 | % change in HAMD response after 6 weeks | Kato et al. (2008) | 0.01 | |
C3435T | rs1045642 | n.s. | |||||
C1236T | rs1128503 | n.s. | |||||
Haplotype 3435C-2677G-1236T | 0.014
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||||||



{kind=link}