Chapter 10 The Spectrum of Epilepsies Associated with Generalized Spike-and-Wave Patterns
Introduction
The generic term epilepsy embraces a particular group of paroxysmal brain disorders, but by itself is of limited clinical value. Intense clinical and genetic research over the last few decades has identified a large number of well-defined epilepsy syndromes with different clinical, EEG, neuropsychological, and neuroimaging profiles, natural history and prognosis, and conditions that ultimately require different management. There is evidence that early treatment can reduce the risk of seizure recurrence,1 and its efficacy depends largely on the appropriate drug choice in relation to the particular clinical syndrome.2 Therefore, identification of the particular form or syndrome is the cornerstone of meaningful, optimal management of the individual patient.2,3
THE INTERNATIONAL CLASSIFICATION OF EPILEPTIC SEIZURES AND SYNDROMES: BASIC CONCEPTS AND DEFINITIONS
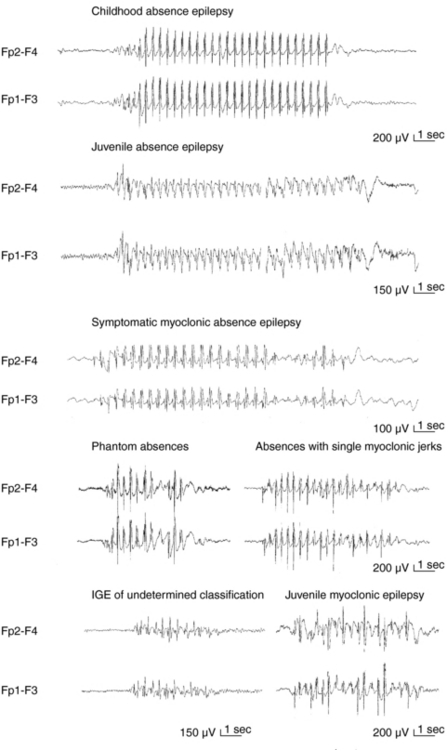
Apart from assisting patients’ clinical management and prognostication, recognition of different syndromes enables effective communication between clinicians and clinical and genetic research. The current classification of epileptic seizures4 and epilepsies5 of the International League Against Epilepsy (ILAE) and also the new ILAE proposal6,7 are essentially organized around two dichotomies in terms of their topography between generalized and localized (partial or focal) and in terms of their etiology between idiopathic epilepsies (genetically determined) and symptomatic epilepsies (due to cerebral pathology). The ILAE classification is currently under revision, but is still valid. Presumed symptomatic epilepsies, in which no certain etiology can be demonstrated, are sometimes referred to as “cryptogenic.” Generalized epilepsies manifest with generalized seizures, whose first ictal clinical changes reflect involvement of both hemispheres and in which initial EEG changes are bilateral, as, for example, the typical absences (TA) that are associated with 3-Hz GSW (Figure 10-1). Localization-related epilepsies (also called focal or partial) manifest with focal (or partial) seizures, in which the initial activation of a group of neurons is limited to a part of one hemisphere5; the EEG shows focal abnormalities, but GSW discharges may occur as a result of secondary bilateral synchrony (SBS).8
CONVENTIONS AND LIMITATIONS OF THE ILAE CLASSIFICATION SYSTEM AND SYNDROMIC DIAGNOSIS IN CLINICAL PRACTICE
Identification of the seizure type(s) is the quintessential element of an epileptic syndrome and the first step for its definition.2 However, it is not always straightforward, particularly on historical evidence (accounts of patients and witnesses) only. TA in idiopathic generalized epilepsy (IGE), for example, may share some principal clinical features with limbic complex partial seizures (CPS) (such as unresponsiveness and automatisms), and one must turn to either their onset (presence or absence of aura) or immediately post-termination (presence or absence of confusion) to make an educated clinical hypothesis. Interictal EEG studies will assist diagnosis by showing focal epileptiform activity or GSW, but even so the rate of misdiagnosis is high with the generalized epilepsies being erroneously diagnosed as focal rather than the converse.9,10 Important reasons for such misdiagnosis include usually short-lived “focal” clinical symptoms and signs, such as unilateral jerks and rotatory seizures in juvenile myoclonic epilepsy (JME),10–13 versive absences,14 and in addition, well-recognized asymmetrical or focal interictal EEG changes.11,15,16 Such electroclinical focalities within the generalized seizures are interpretable within the model of generalized corticoreticular epilepsy, but stand uncomfortably within the inflexible and simplified generalized versus focal dichotomy of the ILAE seizure and syndrome classification system.17 Focal epilepsies may (also) present with rapidly generalized convulsions and infrequently with interictal GSW discharges as in SBS.
In this chapter, we will discuss primarily the various syndromes of IGE, whose GSW is the defining electrographic trait. From the brief foreword, it follows that GSW discharges do not always indicate IGE or indeed generalized epilepsies, and Table 10-1 includes all the main epilepsies and syndromes that are associated with 3–4 Hz GSW. The interictal EEG alone cannot be used for establishing or excluding the diagnosis of epilepsy (including IGEs) or provide oversimplified clues for reliable syndromic diagnosis; this can only be based on a comprehensive electroclinical synthesis for the individual patient, without which EEG interpretation may be completely meaningless and even misleading.2,17
TABLE 10–1 Epilepsy Syndromes and Conditions Associated with GSW Discharges
| Seizures Types | Interictal EEG | |
|---|---|---|
| IGE | TA, MS, GTCS, rarely tonic | Normal background 2.5–4 Hz regular GSW, nonlocalizing focal |
| Absence syndromes | TA, late GTCS, rarely MS (in JAE) | |
| CAE, JAE | ||
| Phantom TA | Brief TA, GTCS; frequent ASE | |
| Myoclonic syndromes | 3-6 Hz irregular GPSW with fragmentations | |
| BMEI | MS | |
| JME | MS, GTCS, TA (35%) | |
| EMA/PMA | TA, GTCS, rarely MS and absence status epilepticus | |
| GTCS only | GTCS | GSW at 2.5–6Hz, regular or irregular ± fragmentations |
| Mixed/unclassified | TA, MS, GTCS, tonicabsences | |
| Generalized Cryptogenic/Symptomatic Epilepsies | ||
| Focal Symptomatic/Cryptogenic Epilepsies | ||
| Various syndromes of lobar (mainly frontal and temporal lobe) epilepsies | Simple or complex partial with semiology reflecting the lobe of origin or propagation | Focal slowing/spikes; may show secondary bilateral synchrony |
| Focal Idiopathic Epilepsies | ||
| Benign rolandic epilepsy | Rolandic seizures, rare brief TA | Normal background, multifocal, rolandic or occipital spikes, GSW in both rolandic and Panayiotopoulos Syndrome |
| Panayiotopoulos syndrome | Autonomic seizures ± eye deviation and motor symptoms. Rolandic and autonomic seizures may coexist in mixed phenotypes; rare TA | |
| Epileptic encephalopathy with CSWS | Generalized and partial seizures mainly during sleep and typical or atypical absences during awake | Continuous SW activity during nnn-RFM sleep (electrical status epilepticus) |
| De Novo Absence-like Status | Unremarkable background without interictal spikes; CSW during status | |
| None | ||
IGE
Notwithstanding the aforementioned diagnostic limitations and pitfalls, GSW discharges are the characteristic EEG building blocks of all seizures in IGE that include typical absences (TA), myoclonic seizures (MS), and GTCS. EEG background activity is normal. IGE comprise a group of genetically determined epilepsies, unrelated to any structural brain pathology, and associated with normal neurological and neuropsychological status. Suitable AED include valproic acid VPA (arguably the first choice), clonazepam (CLZ), ethosuximide (ESX), lamotrigine (LTG), levetiracetam (LEV), topiramate (TPM), and zonisamide (ZNS) (with specific indications against main seizure types),18–20 whereas carbamazepine (CBZ), oxcarbazepine (OCBZ), vigabatrin (VGB), tiagabine (TGB), gabapentin (GBP), pregabalin (PGB), and phenytoin (PHT) are generally contraindicated, as they can cause seizure worsening.2,20–23
BENIGN MYOCLONIC EPILEPSY IN INFANCY (BMEI)2,24–26
This is the earliest form of IGE with a prevalence of about 1% to 2% of epilepsies that start before the age of 4 years. MS start between 4 months to 3 to 4 years and are typically favored by sleep. Two-thirds of the affected children are boys. MS are frequently described by parents as “spasms” or “head-nodding” attacks, and indeed differentiation from spasms may be difficult from description, so ictal video EEG recordings with dedicated EMG electrodes have to be employed. MS are associated with brief bursts of GSW, but interictal EEG is normal. MS may be provoked by photic stimulation in 20% and by unexpected acoustic or tactile stimuli in 10% of children; the latter may be easier to control. Simple febrile seizures occur in 10%, but there are no other seizure types. BMEI responds to VPA, and remission usually occurs within 1 year from onset, but 10 to 20% may develop infrequent GTCS in their early teens. Differential diagnosis includes nonepileptic conditions, such as hypnagogic jerks and benign nonepileptic myoclonus and epileptic syndromes, including severe myoclonic epilepsy of infancy Dravet syndrome), familial infantile myoclonic epilepsy (mapped to 16p13), infantile spasms (West syndrome), epilepsy with myoclonic-astatic seizures (MAE), and the myoclonic form of Lennox-Gastaut syndrome (LGS).
EPILEPSY WITH MYOCLONIC-ASTATIC SEIZURES (MAE)2,27,28
MAE is a genetically determined (some patients fall into the spectrum of generalized epilepsy with febrile seizures plus [GEFS+] nonlesional generalized epilepsy that affects previously normal children between 2 and 5 years of age, but has variable course ranging from good responsiveness to treatment, with normal cognitive functions and even remission, to nonprogressive epileptic encephalopathy with seizure intractability and cognitive impairment. Therefore it clinically overlaps with both IGE (see also the 2001 ILAE proposal, where it features within the IGE6) and cryptogenic/symptomatic generalized epilepsies (see relevant following section). Prevalence is around 1to 2% of all childhood epilepsies, onset ranges between 7 months and 6 years, peaking between 2 and 4 years, and two-thirds of patients are boys. Myoclonic-astatic seizures are the defining seizure symptom and consist of a myoclonic jerk immediately followed by loss of muscle tone. Either component can cause falls, head nodding, or bending of the knees. More than half of patients have brief absences, often with myoclonic jerks, facial myoclonias, and atonia. In two-thirds of patients, febrile and nonfebrile GTCS precede myoclonic astatic seizures by several months. Episodes of absence status may occur, usually introduced by inappropriate treatment such as CBZ; they are frequent in the cryptogenic form. Myoclonic-atonic seizures are associated with discharges of irregular GSW or polyspike waves at >2.5 Hz, with the atonic part of the seizure corresponding to the slow wave of the GSW complex and being associated with diffuse electromyography (EMG) paucity. VPA at high doses is the first drug of choice, combined if needed with LEV, ESX, CLZ, or sulthiame. MAE shares many clinical features with cryptogenic Lennox-Gastaut syndrome, particularly its myoclonic form.29
CHILDHOOD ABSENCE EPILEPSY (CAE)2,30–32
TA are associated with severe impairment of consciousness (unresponsiveness and interruption of ongoing activities) and are usually multiple per day, hence the term pyknolepsy. They have abrupt onset and termination and last from 4 to 20 sec (mainly around 10 sec). Automatisms occur in two-thirds of the seizures but are not stereotyped. Mild myoclonic elements of eyes, eyebrows, and eyelids may feature at the onset of an absence. More severe and sustained myoclonic jerks of facial ro limb muscles indicate other IGEs. TA are nearly always provoked by hyperventilation, even in the clinic. Other types of seizures are incompatible with CAE except for febrile seizures and solitary or infrequent GTCS (usually in adolescence after absences have remitted).
The interictal EEG may show characteristic rhythmic posterior delta activity and brief, usually regular GSW. Monotherapy with VPA ESX, or LTG is successful in most cases, and remission occurs before the age of 12 years with less than 10% of patients developing infrequent GTCS in adolescence or adult life.33 As in all IGEs, VGB, TGB, CBZ, and GBP can produce worsening of absences. CAE is not the only IGE absence syndrome in the first decade of life, and meticulous differentiation from other absence syndromes of worse prognosis is clinically important.
EPILEPSY WITH MYOCLONIC ABSENCES (E-MA)34,35
This rare type of generalized epilepsy was described by Tassinari36,37 and is characterized by myoclonic absences that are associated with 3-Hz regular GSW activity like the typical absences in childhood or juvenile absence epilepsies, but also with bilateral myoclonic jerks of the arms and legs that are time locked to the spike component of the ictal discharge and have a superimposed tonic contraction resulting in a characteristic stepwise upward abduction of the arms. Other seizure types may include GTCS, clonic seizures, and rarely simple absences without myoclonus. It affects less than 1% of children with epileptic disorders, mostly boys (70%), between 5 months and 13 years (median 7 years). Etiology is heterogeneous, and only one-third of patients are of normal neurological and mental state (idiopathic form). These retain normal EEG background. The others are symptomatic, with learning difficulties manifested before or after seizure onset. Early control of absences may secure normal development. The main task of differential diagnosis here is to distinguish between idiopathic and symptomatic cases (see later discussion).
Related posts:
 The Life-Threatening Epilepsies of Childhood and Their Treatment
Psychosis of Epilepsy
The Life-Threatening Epilepsies of Childhood and Their Treatment
Psychosis of Epilepsy
 The Surgery of Temporal Lobe Epilepsy I—Historical Development, Patient Selection, and Seizure Outcome
The Surgery of Temporal Lobe Epilepsy I—Historical Development, Patient Selection, and Seizure Outcome
 Brain Stimulation in Epilepsy—An Old Technique with a New Promise?
Brain Stimulation in Epilepsy—An Old Technique with a New Promise?
 Cortical Myoclonus and Epilepsy: Overlap and Differences
Cortical Myoclonus and Epilepsy: Overlap and Differences
 Does Early Treatment Influence the Long-Term Outcome of Epilepsy?
Does Early Treatment Influence the Long-Term Outcome of Epilepsy?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


