Chapter 11 TIAs and Strokes
Transient Ischemic Attacks
Carotid Artery TIAs
Platelet emboli that form on plaques at the common carotid artery bifurcation (Fig. 11-1) can lead to cerebral-hemisphere TIAs. Contralateral hemiparesis, hemisensory loss, paresthesias, or homonymous hemianopsia characterize carotid artery TIAs. Depending on whether the dominant or nondominant carotid artery gives rise to the problem, a TIA may induce neuropsychologic aberrations accompanied or unaccompanied by hemiparesis and other physical deficits. For example, dominant-hemisphere TIAs may suddenly, but briefly, cause aphasia with or without hemiparesis or homonymous hemianopsia. Similarly, nondominant-hemisphere TIAs may cause brief neglect or hemi-inattention with or without homonymous hemiparesis.
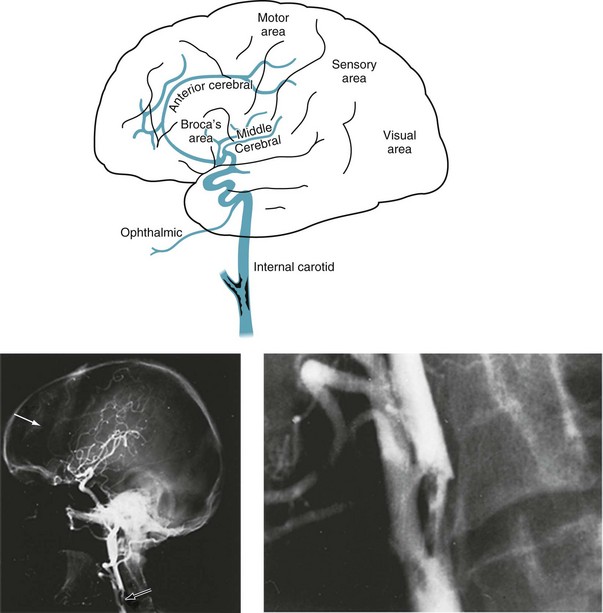
FIGURE 11-1 Top, At its bifurcation in the neck, the common carotid artery divides to form the external and internal carotid arteries. Giving off no branches until it is within the skull, the internal carotid artery immediately sends off the ophthalmic artery. The internal carotid artery then divides into the anterior and middle cerebral arteries. It also gives rise to the posterior communicating artery – not the posterior cerebral artery. Thus, each internal carotid artery perfuses the ipsilateral eye and anterior and middle portions of the ipsilateral cerebral hemisphere. The middle cerebral artery, a major branch of the internal carotid artery, supplies the deep and mid-section of the hemisphere. This region contains most of the motor cortex, sensory cortex, and, in the dominant hemisphere, the perisylvian language arc (see Fig. 8-1). Each anterior cerebral artery, another branch of the internal carotid artery, supplies the frontal lobe, including the medial surface of the motor cortex, which contains the motor innervation for the leg. The posterior cerebral artery (see Fig. 11-2), terminal branches of the basilar artery, supplies the occipital lobe, which houses the visual cortex, and most of the temporal lobe. Bottom left, An arteriogram of the carotid artery and its branches prominently displays the bifurcation (black arrow), the typical “candelabra” of branches that comprise the middle cerebral artery, and a faint anterior cerebral artery that sweeps from anterior to posterior (white arrow). Bottom right, A magnification of the bifurcation reveals an extensive circumferential plaque constricting the internal carotid artery. The remaining blood flow appears as an “apple core.” The rough interior surface of the artery gives rise to retinal and cerebral emboli.
Sometimes a TIA causes only brief visual loss in one eye, i.e., monocular blindness. Neurologists call this distinctive symptom amaurosis fugax (Greek, fleeting darkness). The underlying mechanism consists of emboli from the internal carotid artery flying into the ophthalmic artery, the carotid artery’s first branch, to induce several minutes of ischemia in the retina and optic nerve (Box 11-1). Unlike migraines, which can also cause transient visual loss, TIAs rarely cause headache or scintillations.
In patients with mild cognitive impairment, TIAs may produce a brief but marked confusional state. A similar disturbance may occur after atherosclerosis slowly occludes one carotid artery, forcing the other carotid artery to supply both cerebral hemispheres through the circle of Willis (Fig. 11-2, top right). In this case, the cerebral blood supply is tenuous and emboli from the patent artery produce bilateral cerebral ischemia.
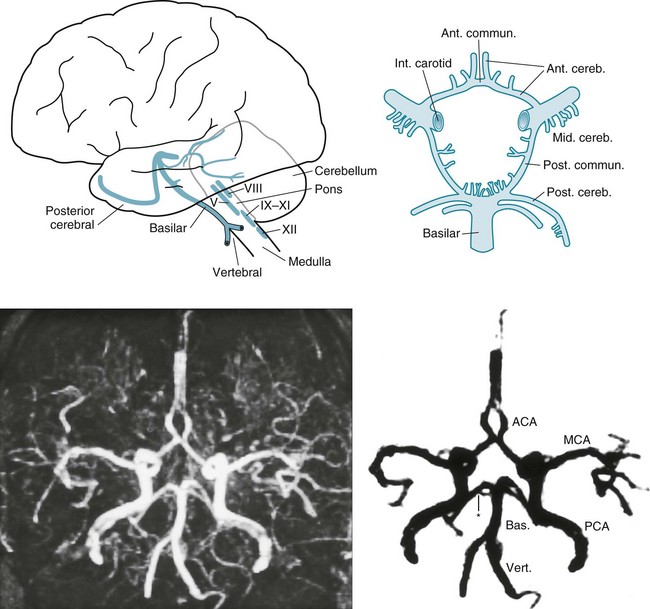
FIGURE 11-2 Top left, After ascending, encased in the cervical vertebrae, the two vertebral arteries enter the skull. They join to form the basilar artery at the base of the brain. Small, delicate branches of the basilar artery supply the brainstem and its contents. (The Roman numerals refer to cranial nerve nuclei.) Large branches, as if wrapping their arms around the brainstem, supply the cerebellum and posterior portion of the cerebrum (i.e., the occipital lobes and inferomedial portions of the temporal lobes). The posterior cerebral arteries are, for practical purposes, the terminal branches of the basilar artery. They supply the occipital cortex and the posterior, inferior aspect of the temporal lobes. Top right, Belying its name, the circle of Willis, the “great anastomoses,” is completely patent in only about 20% of people. Connections between the basilar and internal carotid arteries ideally form the circle, which should give off the anterior, middle, and posterior cerebral arteries. The circle should provide anastomoses between anterior–posterior and right–left cerebral circulations. Despite the advantages that the circle confers, junctions of the arteries are weak spots. Defects at the junctions may balloon outward to form berry aneurysms that rupture and produce subarachnoid hemorrhages. Bottom left, This axial magnetic resonance angiogram (MRA) shows the major cerebral arteries that form the circle of Willis and anterior and posterior circulations. Bottom right, This MRA highlights the major cerebral arteries and shows the anterior and posterior circulations. The anterior circulation consists of the middle cerebral arteries (MCA), anterior cerebral arteries (ACA), and the anterior communicating artery (∧), which joins the ACAs. The posterior circulation consists of the basilar (Bas.) artery and two vertebral (Vert.), posterior cerebral (PCA), and posterior communicating arteries (*), which connect the PCAs and the basilar artery.
Preventive Measures
For patients who cannot undergo carotid endarterectomy, an alternative is intravascular insertion of stents. These devices are essentially expandable tubes that neuroradiologists insert intravascularly into the carotid artery to widen atherosclerotic stenoses (see Fig. 20-28). They also trap underlying atheromatous debris against the inner surface of the arterial wall to reduce the likelihood of emboli. In contrast to the benefits of inserting stents into the extracranial carotid arteries, inserting stents into intracerebral arteries, such as the basilar or middle cerebral artery, carries too much risk to justify its use.
Basilar Artery TIAs
The vertebrobasilar system, basilar artery system, or simply the posterior circulation supplies the brainstem, cerebellum, and the posterior inferior portion of the cerebrum (the occipital and medial inferior portion of the temporal lobes [see Fig. 11-2]). Emboli-generating plaques tend to develop at both the origin of the vertebral arteries (in the chest) and their junction at the base of the brain.
Symptoms and signs of basilar artery TIAs, which usually result from patchy brainstem ischemia, differ greatly from those of carotid artery TIAs (Box 11-2). Typical basilar artery TIA symptoms include tingling around the mouth (circumoral paresthesias), dysarthria, nystagmus, diplopia, ataxia, and vertigo. On rare occasions, when all blood flow through the basilar artery momentarily stops, almost the entire brainstem suffers from ischemia. The brainstem ischemia interrupts consciousness and body tone, which causes patients to collapse. This TIA, termed a drop attack, strikes suddenly and unexpectedly. (It appears similar to cataplexy [see Chapter 17].)
Transient Global Amnesia
TIAs sometimes impair the circulation of the basilar artery’s terminal branches, the posterior cerebral arteries, which supply the temporal lobes (see Fig. 11-2). Because the temporal lobes contain portions of the limbic system, particularly the hippocampi (see Fig. 16-5), posterior circulation TIAs may induce episodes of temporary amnesia and personality change – called transient global amnesia (TGA).
Even in the absence of a confirmatory laboratory test for TGA, its clinical features differentiate it from other neurologic conditions. TGA patients’ preserved intellect and general knowledge, as well as their remaining fully conscious, distinguishes them from patients in delirium. TGA patients, despite their amnesia, also do not confabulate in the manner of Wernicke–Korsakoff patients. Complex partial seizures differ by producing dulling of the sensorium, simple repetitive actions, paroxysmal or other epileptiform EEG changes, and a high rate of recurrence (see Chapter 10).
Strokes
Risk Factors
Reflecting the necessity of preventing strokes, epidemiologic studies constantly search for stroke risk factors (Box 11-3). Although physicians customarily list stroke risk factors as individual threats, the factors actually tend to cluster. For example, obesity, elevated low-density lipoprotein cholesterol, and hypertension frequently occur together in the metabolic syndrome. Some factors, by themselves, have a weak correlation with stroke, but pose a synergistic risk when paired with others. For example, cigarette smoking correlates with stroke; however, with concomitant use of oral contraceptives or in sufferers of migraine with aura, it becomes a moderately powerful risk factor. More strikingly, smokers who are hypertensive have a 20-fold greater risk. Curiously, total serum cholesterol carries a strong risk for myocardial infarctions but a weak risk for stroke.
Elevated serum concentration of homocysteine serves as a marker for an increased risk. In the autosomal recessive genetic condition, homocystinuria, children have a Marfan-like habitus, ocular lens displacement, and other anatomic abnormalities. More importantly, they routinely suffer strokes in childhood. Elevated homocysteine levels in adults are associated with the use of antiepileptic drugs and coronary artery and peripheral vascular disease, as well as stroke. Folic acid reduces elevated serum homocysteine concentrations (see Fig. 5-8), as well as reducing the incidence of fetal neural tube defects (see Chapter 13), but surprisingly, it does not reduce the incidence of strokes.
Thrombosis and Embolus
In addition to creating permanent clinical deficits, stroke scars are potentially epileptogenic. Approximately 50% of seizures that develop in adults older than 65 years originate from these lesions (see Chapter 10). Thus, patients with a history of having sustained a stroke who develop confusion, unresponsiveness, or abnormal behavior may have suffered a complex partial seizure as well as another stroke or TIA.
Necrosis
Infarctions in the Carotid Artery Distribution
Cerebral artery thrombosis and embolism produce an infarction in the distribution of the occluded artery. These strokes result in well-known clinical deficits (see Fig. 11-1 and Box 11-4):
• Anterior cerebral artery infarction damages the anterior and medial aspects of the frontal lobe. It typically causes paresis and apraxia of the contralateral leg. With bilateral anterior cerebral artery infarctions, the resulting frontal lobe damage, which is extensive, typically causes pseudobulbar palsy, apathy, mutism, personality changes, and urinary incontinence (see Chapter 7), along with both legs having paresis and apraxia.
• Middle cerebral artery infarction, which is the most common stroke, usually results in contralateral hemiparesis, hemisensory loss, and either aphasia with dominant hemisphere lesions or hemi-inattention, including neglect, with nondominant hemisphere strokes.
Infarctions in the Basilar Artery Distribution
Infarctions in the basilar artery distribution cause brainstem, cerebellar, or posterior cerebral injuries. Small brainstem infarctions usually cause constellations of cranial nerve injuries and hemiparesis, and large ones usually cause coma, if not immediate death. In contrast to cerebral-hemisphere infarctions, brainstem infarctions generally spare language or intellectual function and do not cause seizures (see Box 11-4).
The posterior cerebral arteries, as previously noted, are actually terminal branches of the basilar artery. Infarction of a posterior cerebral artery causes a contralateral homonymous hemianopsia and occasionally alexia without agraphia (see Chapter 8). Bilateral posterior cerebral artery strokes can cause (cortical) blindness, which sometimes leads to Anton syndrome (see Chapter 12).
Neurologists love to localize small brainstem infarctions for both clinical and academic reasons. Lesions of the midbrain cause ipsilateral oculomotor nerve and contralateral paresis (see Fig. 4-9). Those in the pons cause ipsilateral abducens nerve and contralateral paresis (see Fig. 4-11). Infarctions involving the midline of the midbrain or pons cause the medial longitudinal fasciculus syndrome (see Chapters 12 and 15). Finally, lateral medullary infarctions, which are the most common brainstem infarction, cause a complex, apparently disparate, combination of ipsilateral limb ataxia, palatal paresis, Horner syndrome, and alternating hypalgesia (see Wallenberg syndrome, Fig. 2-10).
Hemorrhages
Hemorrhages, which are most often the result of hypertension, usually erupt in the basal ganglia, thalamus, pons, or cerebellum (see Figs 20-13 and 20-14). Trauma and use of cocaine also cause hemorrhages, but their location is not as predictable as with hypertension-induced ones. In a classic problem, patients who take a monamine oxidase inhibitor (MAOI) antidepressant and then, inadvertently or in a suicide attempt, consume certain foodstuffs, notoriously aged cheese or red wine, or receive meperidine (Demerol), develop cerebral hemorrhages. These combinations, which are sympathomimetic, lead to a burst of hypertension sufficient to cause the hemorrhage. (Notably, MAOI-A antidepressants may cause this problem, but MAOI-B medications, such as those used to treat Parkinson disease, generally do not [see Chapter 9].)
Subarachnoid hemorrhage (SAH), although usually traumatic in origin, sometimes results from a ruptured berry aneurysm. SAH most often produces a prostrating headache that patients classically describe as the “worst headache” of their life. SAH also produces nuchal rigidity and lethargy, but usually no lateralized signs. Exercising, straining at stool, and other usually benign activities can rupture an aneurysm and precipitate an SAH. A diagnostic dilemma occurs when a sudden, profound headache interrupts sex. In this case, the exertion may have led to an SAH, but more often coital cephalalgia is responsible (see Chapter 9). Thus, TGA, SAH, and a migraine variant are hazards of sexual activity.
Neuropsychologic Sequalae
Several large or many small (lacunar) strokes cause vascular cognitive impairment, previously labeled vascular dementia, and before that multi-infarct dementia. The preliminary version of the DSM-5 labels it Vascular Neurocognitive Disorder. Ironically, despite the implication of these terms, the dementia stems from the neuropathologic changes of Alzheimer disease that accompany those of cerebral vascular disease (see Chapter 7).
Occlusion of a cerebral artery typically destroys a discrete critical area of the cerebral cortex. Commonly occurring strokes produce well-known neuropsychologic syndromes, such as aphasia, Gerstmann syndrome, and hemi-inattention (see Chapter 8). Strokes of both frontal lobes, which may occur either simultaneously or in succession, cause pseudobulbar palsy and frontal lobe dysfunction (see Chapters 4 and 8).
In another mechanism, hypoxia, hypotension, carbon monoxide poisoning, or similar catastrophes cause generalized cerebral anoxia. This insult leads to dementia, cortical blindness, a persistent vegetative state (PVS: see later), and postanoxic myoclonus (see Chapter 18). A variation of generalized anoxia occurs when cerebral anoxia affects only the fine, terminal branches of cerebral arteries that perfuse the extensive circumferential regions of the cerebral cortex, which neurologists call the watershed or borderzone area. An episode of cerebral anoxia that reduces the oxygen supply below a critical point in the watershed area produces a watershed infarction






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


