Fig. 1
Description of the use of PET brain imaging to determine the outcome measure of receptor availability
2 Imaging β2*-Nicotinic Acetylcholine Receptors
2.1 Preclinical Studies
The nAChRs that contain the β2*-subunit are critical for mediating the effects of nicotine in the brain including the reinforcing effects (Picciotto et al. 1998), dopamine release (Epping-Jordan et al. 1999; Koranda et al. 2013), sensitivity to nicotine (Cosgrove et al. 2010; Marubio et al. 1999; Tritto et al. 2004), and the incentive aspects of motivation (Brunzell et al. 2010) (see chapter entitled The Role of Mesoaccumbens Dopamine in Nicotine Dependence; this volume). In addition, there is a wealth of literature showing that nicotine and tobacco smoking robustly upregulate (i.e., increase numbers of) β2*-nAChRs throughout the brain (Abreu-Villaca et al. 2003; Benwell et al. 1988; Breese et al. 1997; Kassiou et al. 2001; Marks et al. 1992). Preclinical studies administering nicotine at various doses and routes of administration to rats, mice, and monkeys—as well as postmortem human studies—have all indicated that nicotine and tobacco smoke result in significantly more β2*-nAChRs throughout the brain compared to saline (animals) or to not smoking (humans). We now know that nicotine itself is responsible for this upregulation. Nicotine acts in the cell to help the receptor subunits assemble and then acts to chaperone the receptors to the cell membrane (Srinivasan et al. 2010). Our goal was to measure this upregulation in living human tobacco smokers. However, first we needed to work out the proper experimental timing.
Nicotine and the radiotracers used in these studies both bind to the same receptor in the brain—the nAChR containing the β2*-subunit. When nicotine is present in the brain, it may block the receptor and prevent the radiotracer from binding, which would confound our ability to measure β2*-nAChR availability. Our preclinical experiment consisted of two monkeys drinking nicotine (diluted in water and sweetened with Tang to make it more appetizing) for 6 weeks. After 6 weeks, the monkeys were taken off nicotine. One monkey was scanned at 1 day into nicotine withdrawal, and the other was scanned at 2 days into nicotine withdrawal. Surprisingly, the data showed a decrease in radiotracer binding which was not consistent with the literature. To probe further, the monkeys consumed nicotine for two additional weeks and then were scanned at 7 days of withdrawal. At that point, there was a robust increase in radiotracer binding suggestive of an upregulation of β2*-nAChRs that was consistent with the preclinical literature. Taken together, these data suggested that nicotine remains in the brain during early withdrawal and may take up to 7 days to clear. Levels of cotinine (the major metabolite of nicotine) in the monkeys were measured over the 7 days of withdrawal. Cotinine progressively declined over the week, not completely clearing or reaching nonsmoker levels until 7 days of abstinence. The cotinine data nicely mirrored the brain data. Once cotinine had cleared, it was possible to measure nicotine-induced upregulation of β2*-nAChRs in the brain. In the human studies discussed below, low cotinine levels are typically used as an indicator of abstinence and that nicotine has cleared so that smokers can be imaged with β2*-nAChR radiotracers.
2.2 Imaging the Upregulation of β2*-nAChRs in Tobacco Smokers
Based on the preclinical monkey study, our group imaged β2*-nAChRs in human tobacco smokers at 7–9 days of smoking abstinence (early phase withdrawal). In this and similar studies, the subjects were required to quit smoking and not use any medications or nicotine replacement strategies such as the nicotine patch, because all forms of nicotine would bind the β2*-nAChR and block the radiotracer from binding. In order to help the subjects quit smoking, we used contingency management techniques (Staley et al. 2006). In the first paper, we demonstrated that tobacco smokers at 7–9 days of abstinence have significantly higher β2*-nAChR availability in the cortex, striatum, and cerebellum compared to a group of age- and sex-matched nonsmokers (Fig. 2). This work in our laboratory (Staley et al. 2006) and others (Mamede et al. 2007; Mukhin et al. 2008) confirmed that it is possible to measure the upregulation phenomenon in human smokers in vivo.
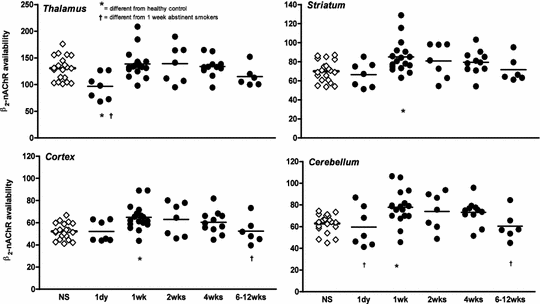
Fig. 2
β2*-nAChR availability (V T /f P ) is shown in individual nonsmokers (open diamonds) and tobacco smokers (filled circles) at 1 day, 1, 2, 4, and 6–12 weeks of abstinence in the thalamus, striatum (average of caudate and putamen), cortex (average of cortical regions including parietal, frontal, anterior cingulate, temporoinsular, and occipital cortex), and cerebellum. The line in each scatter plot represents the mean value of those subjects. Asterisk indicates significant difference from control nonsmokers after Bonferroni’s correction using two-sample t-tests. Dagger indicates significant difference from 1 week abstinent smokers after Bonferroni’s correction using planned post-hoc between-group comparisons subsequent to the analysis of repeated measures mixed-effects regression models including the overall effect of abstinent smoker group
2.3 Imaging the Normalization of β2*-nAChRs in Tobacco Smokers
There is also evidence from preclinical (Collins et al. 1990; Pietila et al. 1998) and postmortem human (Breese et al. 1997) studies that the β2*-nAChRs do not stay upregulated and eventually return to control levels. The postmortem study (Breese et al. 1997) indicated that smokers who had quit smoking at least two months prior to their death had β2*-nAChRs levels similar to controls. However, smokers in the study had quit anywhere from 2 months to 30 years prior to their death, so the study did not shed light on the acute time course of receptor changes (e.g., during acute withdrawal in the first few months of abstinence, when relapse rates are high). In our next study, we imaged β2*-nAChR changes over the first few months of abstinence in tobacco smokers (Cosgrove et al. 2009). As shown in Fig. 2, at one day of abstinence, nicotine is still present in the brain blocking the radioligand from binding to the receptor and there is no difference in β2*-nAChR availability compared to the group of nonsmokers. At one week of abstinence, there is higher β2*-nAChR availability in smokers compared to nonsmokers consistent with the previous study (Staley et al. 2006). Then even at 2 and 4 weeks of abstinence, receptor availability remains high and does not return to nonsmoker control levels until 6–12 weeks of abstinence. This study (Cosgrove et al. 2009) and others (Brody et al. 2013b; Mamede et al. 2007) demonstrate that upregulation of β2*-nAChRs is initially persistent, but that β2*-nAChRs normalize over approximately 6–12 weeks of abstinence from cigarettes and all other nicotine-containing products. These brain changes parallel the clinical course of smoking cessation in which craving, relapse, and withdrawal symptoms slowly dissipate over the first few months of abstinence even though relapse may occur months or years after the last cigarette.
Relationships between β2*-nAChR availability and clinical correlates have been reported in these studies. The type of cigarette smoked modulates the degree of upregulation. Cigarettes containing menthol, which are used by up to 1/3 of smokers, lead to higher β2*-nAChR availability than non-menthol-containing cigarettes (Brody et al. 2013a). Additionally, a primary advantage of neuroreceptor imaging studies (vs. postmortem studies) is that we can record behavior and examine correlations between behaviors of interest and brain chemistry. At one week of abstinence, β2*-nAChR availability in the sensorimotor cortex was negatively correlated with the urge to smoke to relieve withdrawal symptoms (Staley et al. 2006). At four weeks of abstinence subjects with higher β2*-nAChR availability in the cerebellum reported both a greater desire to smoke and a greater urge to smoke to relieve withdrawal (Cosgrove et al. 2009). This suggests that magnitude of upregulation may play a role in craving over the course of abstinence and that managing the time course of the normalization may help individuals who are more likely to relapse in response to high levels of craving. For example, it is possible that nicotine replacement strategies may be effective because they continue to activate β2*-nAChRs. This leads to continued upregulation and the individual can “wean” the receptors off of nicotine as the dose of nicotine is decreased over time.
2.4 Sex Differences in β2*-nAChR Availability
There is a large literature demonstrating sex differences in tobacco smoking behaviors. In general, men tend to smoke for the nicotine reinforcement, or nicotine per se in the cigarette, whereas women tend to smoke more for the sensory cues associated with smoking, as well as affect and stress regulation (Perkins 2009; Perkins et al. 1999; Perkins and Scott 2008). There are also two preclinical studies showing that male rats and mice exposed to nicotine exhibited greater nAChR upregulation than female rats and mice exposed to nicotine (Koylu et al. 1997; Mochizuki et al. 1998). We wanted to determine if there were sex differences in β2*-nAChR availability between men and women smokers compared to nonsmokers. Consistent with the preclinical literature, male smokers had significantly higher β2*-nAChR availability compared to male nonsmokers (9–17 %), but women smokers had similar β2*-nAChR availability compared to women nonsmokers (1–3 %) (Cosgrove et al. 2012). This was a striking finding given in all the studies demonstrating that nicotine and tobacco smoking upregulate β2*-nAChRs throughout the brain. Considering known behavioral sex differences in tobacco smoking, these findings make sense and provide a biological mechanism that may underlie some of the behaviors. Specifically, men smoke for the nicotine in cigarettes, they are more responsive to nicotine replacement therapy as a cessation strategy, and men’s brains are responsive to nicotine, exhibiting upregulation of β2*-nAChRs. Women smoke for affect regulation and for reasons other than the nicotine, they do not respond as well to nicotine replacement strategies, and their brains do not respond to nicotine by increasing β2*-nAChRs. The bottom line is that novel treatment strategies targeting other receptor systems need to be evaluated to more effectively help women quit smoking. All the current strategies act at the β2*-nAChR, and, of course, all nicotine replacement strategies act at that site. For example, varenicline (Chantix) is a partial agonist at the β2*-nAChR, and even bupropion (Zyban) is a nicotinic antagonist.
2.5 Nicotine Occupancy of β2*-nAChRs
In addition to receptor changes, imaging studies have informed our knowledge about what happens in the brain after someone smokes a cigarette. For example, after one puff of a cigarette, approximately 50 % of all β2*-nAChRs in the brain are occupied by nicotine. After smoking one or two cigarettes, the receptors are saturated, so up to 100 % of β2*-nAchRs are occupied by nicotine (Brody et al. 2006a; Esterlis et al. 2013). We know that nicotine doesn’t clear the brain immediately, and in fact dependent smokers have a slower process of nicotine accumulation going into the brain from a cigarette than do nondependent smokers (Rose et al. 2010). Indeed, in one study, habitual smokers did not show evidence of puff-associated spikes in nicotine, but rather a gradual accumulation of nicotine during smoking (Rose et al. 2010). Both of these ideas—rapid accumulation and puff-associated spikes of nicotine—had been proposed to explain the maintenance of tobacco dependence. So with a slow kinetic profile and with most receptors in a smoker occupied by nicotine throughout the day, why do people keep smoking? This brings up some important points about tobacco smoking. People smoke for many different reasons, and nicotine reinforcement is only one component. The reinforcement or pleasure derived from nicotine, like many other drugs of abuse, is necessary in driving the initial phases of drug-seeking behavior. However, as the addiction progresses, many people may continue to smoke in order to avoid withdrawal symptoms and due to the many conditioned cues that have become ingrained, which are a part of the compulsive, repetitive nature of tobacco smoking. Additionally, there are over 4,000 chemical compounds that are produced when a cigarette burns; all of these compounds are in tobacco smoke and are inhaled. Thus, while nicotine is the primary addictive component of tobacco smoke, there are additional compounds such as MAO-A and MAO-B inhibitors that likely play a role.
Other imaging studies have demonstrated that even smoking a denicotinized cigarette, which supposedly has very low nicotine content, leads to occupancy of approximately 20 % of β2*-nAChRs in the brain (Brody et al. 2009). This is similar to the level of occupancy produced by secondhand smoke. Brody and colleagues at UCLA performed an elegant study examining the effect of secondhand smoke on β2*-nAChRs by having subjects sit in a car (the window was down a few inches) with a person smoking, and they reported up to 20 % of β2*-nAChRs were occupied by nicotine in the individual who was just sitting in the car, not smoking (Brody et al. 2012). Interestingly, several states have recently passed laws prohibiting smoking in the car with children under the age of 18.
In terms of treatment, Esterlis and colleagues conducted a proof-of-concept study (Esterlis et al. 2013) to determine whether a nicotine vaccine, 3′-AmNic-rEPA, would reduce the amount of nicotine entering the brain and binding to β2*-nAChRs. Smokers were imaged before and after treatment with the vaccine, and occupancy of intravenously delivered nicotine was measured. Immunization led to a significant ~13 % reduction in nicotine occupancy confirming that vaccines may help smokers quit smoking by reducing the amount of nicotine available to occupy β2*-nAChRs. While this particular vaccine is not available for treatment, this study highlights an innovative paradigm to test future medications.
3 Imaging Dopamine Release in Response to Nicotine and Tobacco Smoking
3.1 Preclinical Microdialysis Studies
Before PET made possible indirect measurement of dopamine release in vivo, the only way to measure dopamine levels in a living brain was via microdialysis. Microdialysis is used primarily in rodents. During a surgery, a probe is placed through the skull into the region of interest (e.g., the nucleus accumbens). After recovery, DA levels can be sampled in the awake, behaving animal typically in response to a drug or a stimulus. Di Chiara and Imperato performed the seminal study showing that drugs abused by humans release DA in the nucleus accumbens of the rat brain (Di Chiara and Imperato 1988). Amphetamine (1.0 mg/kg, SC) raised DA levels over 1,000 % from baseline, whereas ethanol (1.0 g/kg, IP) and nicotine (0.6 mg/kg, SC) raised DA levels to 200 % over baseline levels. This illustrates how powerful DA release can be when it is directly stimulated with amphetamine, which is both a direct DA releaser and DA reuptake inhibitor. Put into context, a similar dose of amphetamine given to a monkey or a human in a PET experiment, which is an indirect measure of change in DA, would result in a 15–30 % change in binding potential (BP) as measured with [11C]raclopride or another D2/3 ligand.
3.2 Imaging Dopamine Release with PET
Striatal dopamine release has been reliably measured using stimulants such as amphetamine or tobacco smoking with radiotracers such as [11C]raclopride PET or [123I]IBZM SPECT (see Laruelle 2000 for review). More recently, [11C]PHNO has been used, since as an agonist, it labels the high-affinity functional dopamine D2/3 receptors (which theoretically makes it more sensitive to changes in DA compared to [11C]raclopride, an antagonist). Further, because it has a higher affinity for D3 versus D2 DA receptors, this allows for regional interpretation of D3 and D2 receptors (Girgis et al. 2011). Specifically, in humans, binding in dorsal striatum is primarily D2, binding in substantia nigra is primarily D3, and binding in globus pallidus is mixed with approximately 65 % D3 versus D2 dopamine receptors (Tziortzi et al. 2011). For all of the dopaminergic radiotracers, drugs such as amphetamine robustly increase synaptic DA. The increased DA competes with the radiotracer to bind at the dopamine receptor; thus, an increase in DA results in a decrease in radiotracer binding compared to baseline (Fig. 3). This allows calculation of the “occupancy” of the receptors by DA or a change in BP and is an indirect measure of DA release based on the “occupancy model” (Laruelle 2000). Although nicotine is a less robust DA releaser compared to amphetamine, there are many studies that have examined nicotine and tobacco smoking-induced DA release in human subjects.
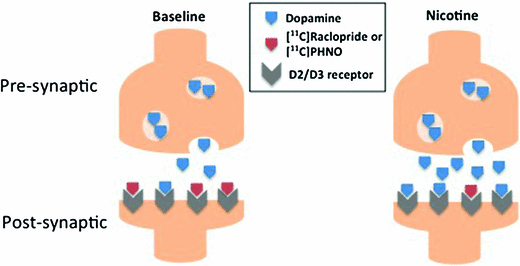
Fig. 3
Binding competition at dopamine D2/D3 receptors between endogenous dopamine and PET radiotracers, [11C]raclopride or [11C]PHNO, at baseline and after nicotine or tobacco smoking. Nicotine increases synaptic dopamine levels, and the dopamine binds to the D2/3 receptors which reduce the number of available binding sites for the radiotracers which also bind the D2/D3 receptors. The change in number of available receptors from the baseline scan to the scan after the drug challenge is an indirect measure of increased dopamine levels
3.3 Imaging Dopamine Release in Smokers
Most of the PET studies of the dopaminergic response to cigarette smoking and/or nicotine are summarized in Table 1. Unless noted, the studies used [11C]raclopride PET imaging. Barrett et al. did one of the earliest studies of smoking with PET; the first study with smokers actually smoking in the scanner (Barrett et al. 2004). While the authors did not find any significant change in BP between the baseline and smoking periods, the subjects who experienced “mood elevating effects in response to smoking” (n = 5) had a 21 % decrease in BP from baseline (i.e., increase in DA) in the caudate. There were some notable innovations in the Barrett et al. study as well as some reasons for caution in interpretation. On the plus side, the smokers smoked in the scanner—thus any DA release detected could be attributed to the entire smoking behavior—something not possible with animals but highly relevant for medications development. The smokers were asked to smoke their own brand—another way of assuring that behavior in the scanner approximated subjects’ smoking behavior. However, the protocol of smoking up to six cigarettes in an hour may have been aversive to some of the subjects and possibly contributed to the variability between subjects.
Table 1
Summary of smoking studies
PET Study | Subjects | Stimulus | Control | Findings |
|---|---|---|---|---|
Brody (2004) | 20 Smokers, 1 scan each | 50 min Post-injection, 10 subjects smoked 1 cigarette at 10-min break outside of scanner; scanned again for an additional 30 min
Related posts: Mechanisms Underlying Nicotine Reinforcement
Disorders as Vulnerability Factors for Nicotine Addiction: What Have We Learned from Animal Models?
Bases of Cue- and Nicotine-induced Reinstatement of Nicotine Seeking: Implications for the Development of Smoking Cessation Medications
Withdrawal Mechanisms Underlying Nicotine Reinforcement
Disorders as Vulnerability Factors for Nicotine Addiction: What Have We Learned from Animal Models?
Bases of Cue- and Nicotine-induced Reinstatement of Nicotine Seeking: Implications for the Development of Smoking Cessation Medications
Withdrawal
 Role of Mesoaccumbens Dopamine in Nicotine Dependence Role of Mesoaccumbens Dopamine in Nicotine Dependence
 Pediatric Spinal Cord Injury Pediatric Spinal Cord Injury
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|