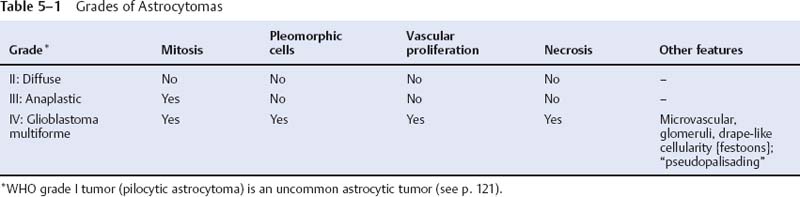
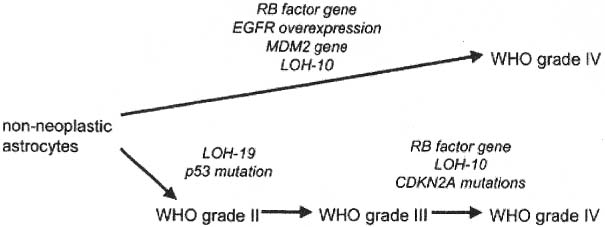
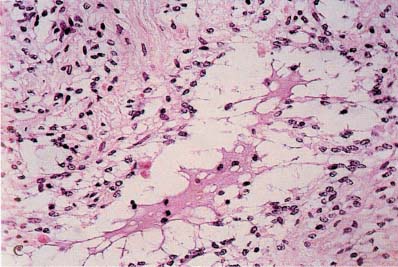
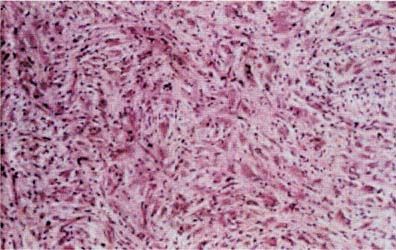
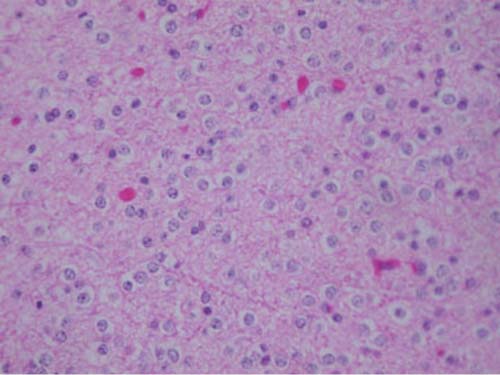
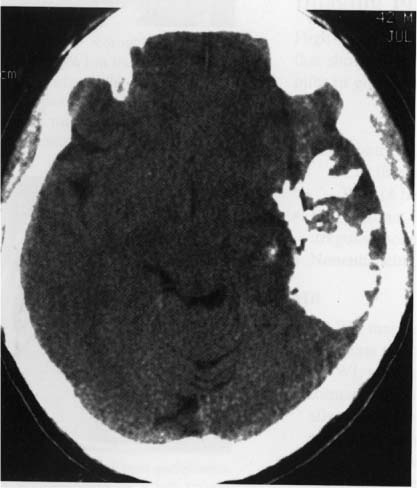
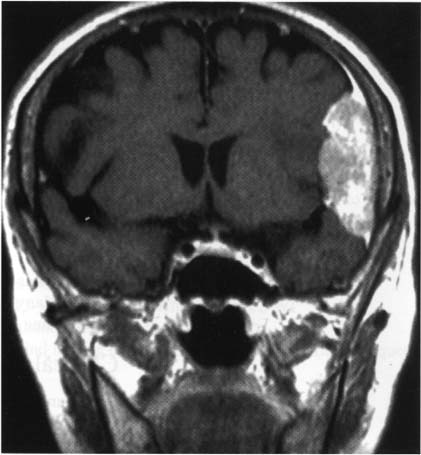
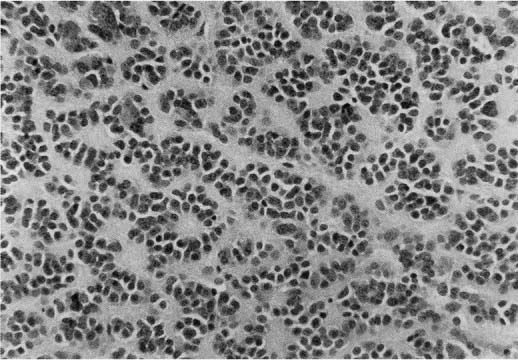
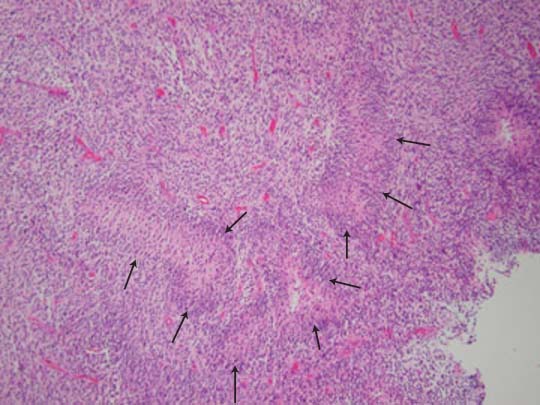
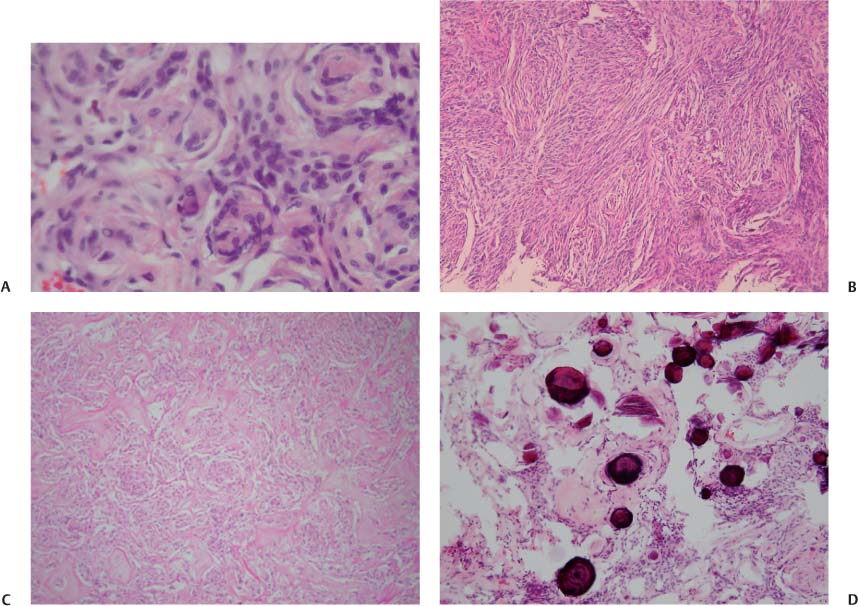
5 Note: Significant diseases are indicated in bold and syndromes in italics. 1. Fibrillary astrocytomas a. tumor grades (Table 5–1) (Fig. 5–1): all exhibit nuclear atypia; can be cytologically defined as malignant according to MIB-1 antibody labeling of the Ki-67 nuclear antigen, which is a protein of unknown function that is expressed only during mitosis (Box 5.1) MIB-1-labeled Ki-67 ratio = The number of labeled cells divided by the number of unlabeled cells b. genetics (Fig. 5–2) i. mutations of the p53 gene, which encodes a cell-cycle regulator that inhibits progression through G1 phase of mitosis; uncommon in primary glioblastoma ii. mutations of the MDM2 gene, which encodes a protein that binds p53 protein (1) mutations may cause overexpression of MDM2 protein, which leads to tumor formation by reducing levels of p53 (2) MDM2 protein also binds mutant p53, thus functional loss of MDM2 may allow abnormally active p53 to cause tumor formation iii. mutations of the epidermal growth factor (EGF) receptor gene, which encodes the receptor for EGF and tumor growth factor (TGF)-α (1) mutations may cause overexpression of mutant EGF receptor that are constitutively active (i.e., do not require ligand binding) (2) even without mutations, wild-type EGF receptor and related receptors (i.e., fibroblast growth factor receptor, vascular endothelial growth factor receptor) are commonly overexpressed in low-grade astrocytomas iv. platelet-derived growth factor-α (PDGF), which is involved in cell proliferation Figure 5–1 Glioblastoma multiforme. Arrows identify pseudopalisading and festoons. v. mutations in the retinoblastoma (RB) factor gene (Box 5.2), which encodes an inhibitory binding protein for transcription factor E2F that promotes proliferation; mutations of RB factor allows inappropriate release of active E2F and progression through G1 phase of mitosis vi. mutations of the CDKN2A (Box 5.3), which encode protein kinases that inhibit progression through the cell cycle by affecting the function of RB and p53 vii. loss of heterozygosity (LOH) on chromosomes 19 and 10: deletion of an unknown gene or genes on one pair of the chromosomes (1) LOH 10 region includes (a) the leucine-rich glioma-inactivated (LGI-1) protein (Box 5.4) (b) PTEN, a tumor suppressor gene Tumors with High “Seeding” Potential c. tumor location: supratentorial >> infratentorial, rarely cerebellum or spinal cord; may be multifocal i. tumors grow along white matter tracts and vasculature, occasionally disseminating into the CSF {seeding}, particularly with tumors in the cerebellum, corpus callosum, or along the ventricles (Box 5.5) d. epidemiology: accounts for 60% of primary brain tumors in adults; only known risk factor is brain irradiation Figure 5–2 Genetic abnormalities of astrocytomas. (EGFR, epidermal growth factor receptor; LOH, loss of heterozygosity; RB, retinoblastoma.) i. grade II/diffuse astrocytoma: observation, or treatment with irradiation or chemotherapy Figure 5–3 Juvenile pilocytic astrocytoma exhibits alternating compact and spongy areas that give a “floating” appearance to some of the cells. Multinucleated giant cells are also seen. (From Keating R et al. Tumors of the Pediatric Central Nervous System. Stuttgart, Germany: Georg Thieme; 2001:53, Fig. 5–6c. Reprinted by permission.) ii. grade III/anaplastic astrocytoma: surgery + irradiation ± chemotherapy iii. grade IV/glioblastoma multiforme: surgery chemotherapy (temozolomide) + irradiation f. prognosis i. grade II/diffuse astrocytoma: 5-year survival ii. grade III/anaplastic astrocytoma: 2.5-year survival iii. grade IV/glioblastoma multiforme: < 1-year survival 2. Uncommon types of astrocytomas a. juvenile pilocytic astrocytoma—grade I astrocytoma of the WHO classification i. histology: tumor exhibits a dense cellular areas filled with long bipolar astrocytes and numerous Rosenthal fibers, which are interspersed with relatively cell-free regions (Fig. 5–3); also exhibits eosinophilic granular bodies (1) forms a large cystic mass when located in the cerebellum but a solid tumors when located elsewhere (2) strongly associated with neurofibromatosis I ii. tumor location: cerebellum > hypothalamus, optic nerve iii. epidemiology: common only in children iv. treatment: surgery is usually curative; tumors in the optic-hypothalamic area respond to chemotherapy with carboplatin and vincristine b. subependymal giant cell astrocytoma i. histology: tumor typically consists of giant astrocytes mixed with differentiated neuron-like cells in a heavily calcified stroma (Fig. 5–4) (1) no anaplastic potential although exhibits locally invasive growth (2) typically grow along ependymal surface of lateral ventricles, may block cerebrospinal fluid flow causing hydrocephalus (3) associated with (a) soft-tissue sarcomas and breast cancer {Li-Fraumeni syndrome} (b) multiple intestinal polyps {Turcot syndrome} (c) phakomatoses: tuberous sclerosis, neurofibromatosis-1 (see Chapter 12) ii. treatment: observation; surgery if the tumor grows or causes hydrocephalus Figure 5–4 Subependymal giant cell astrocytoma. (From Keating R et al. Tumors of the Pediatric Central Nervous System. Stuttgart, Germany: Georg Thieme; 2001:330, Fig. 23–3. Reprinted by permission.) c. pleomorphic xanthoastrocytoma i. histology: tumor exhibits pleomorphic astrocytes (therefore is glial fibrillary acidic protein- [GFAP] positive) filled with lipid vacuoles like adipocytes, but that are rarely mitotic; exhibits a dense reticulin network and a lymphocytic infiltrate, and can be cystic (1) very benign growth (rarely degenerate into malignant tumors), and only are symptomatic when they cause seizures ii. epidemiology: common in children iii. tumor location: cortex (particularly temporal lobe), often extending into the subarachnoid space iv. treatment: surgery is usually curative 1. Histology: composed of small, round cells that are GFAP-negative and that develop from oligodendrocytes Figure 5–5 Oligodendroglioma histology. (Courtesy of Dr. C. Yamada) a. cells exhibit “fried egg” artifact and “chicken wire” vasculature after formalin fixation (Fig. 5–5), resembling the non-artifactual appearance of clear cell ependymomas or central neurocytomas b. 50% of oligodendrogliomas have a mixed histology with pronounced astrocytoma features {oligoastrocytoma} that may ultimately degenerate into a malignant astrocytoma i. both oligodendrocyte and astrocyte components show evidence of malignancy in oligoastrocytomas, that is, astrocytes are not simply trapped in an oligodendroglioma c. frequently are calcified, or have hemorrhagic or cystic components d. tumor location: often developing at the gray–white junction and growing toward the cortex, particularly in the frontal lobe 2. Tumor grades: histologically defined as low grade or anaplastic by the presence of mitosis, pleomorphism, vascular proliferation, and necrosis, as per astrocytomas; cytologically defined as malignant according to MIB-1 antibody labeling, as per astrocytomas 3. Genetics a. p53 gene mutations b. epidermal growth factor, platelet-derived growth factor, and vascular endothelial growth factor overexpression occurs without gene amplification c. loss of heterogeneity (LOH): low-grade oligodendrogliomas have LOH on chromosomes 1 or 19; high-grade oligodendrogliomas also have LOH on chromosomes 9 or 10 (Box 5.6) d. mutations in the CDKN2A gene, which encode the p16 protein cycle regulator (Box 5.7) 4. Tumor location: > frontal temporal, parietal > occipital cortex; location is proportionate to the amount of white matter in these areas 5. Epidemiology: bimodal peak incidences in children 8 years of age and adults 40 years of age 6. Diagnostic testing: Exhibits heterogeneous densities on neuroimaging, most of which are calcified (“chunky” or “popcorn-like” calcification) (Fig. 5–6) 7. Treatment a. surgery ± irradiation (may only observe low-grade oligodendrogliomas after surgery) b. chemotherapy: highly sensitive to PCV treatment (procarbazine, lomustine [CCNU], vincristine) or temozolomide (Temodar), particularly rapidly enlarging tumors with ring enhancement i. response to chemotherapy is not related to MIB-1-labeled Ki-67 ratio but it is proportionate to the amount of oligodendroglial-like cells in the tumor ii. tumors with LOH 1 or 1 + 19 have greater chemotherapy responsiveness Figure 5–6 Oligodendroglioma calcifications. (From Fischbein NJ et al. Teaching Atlas of Brain Imaging. Stuttgart, Germany: Georg Thieme; 2000:10, Fig. A. Reprinted by permission.) a. histologically defined benign oligodendroglioma i. MIB-1-labeled Ki-67 ratio < 5% = 80% 5-year survival ii. MIB-1-labeled Ki-67 ratio > 5% = 25% 5-year survival b. histologically defined anaplastic oligodendroglioma: 20% 5-year-survival overall c. oligoastrocytoma: 60% 5-year survival 1. Histology: a diffuse infiltration of brain parenchyma by neoplastic glial cells, generally occurring in multiple cerebral lobes but occasionally infratentorial a. can develop from glial tumors (20%), but usually develops without preexisting tumor b. exhibits oligodendroglial, astrocytic, or mixed sub-types 2. Symptoms: generally presents with seizures; may have a focal deficit (30%), dementia (30%), or headache (10%) 3. Diagnostic testing: often is nonenhancing on neuroimaging 4. Treatment: depends upon histology but typically treated with irradiation ± chemotherapy 5. Prognosis: 12-month-survival untreated, 2.5-year-survival treated a. patients with oligodendroglial subtype have a significantly longer survival 1. Histology (Fig. 5–7): the MIB-1 antibody labeling of the Ki-67 antigen can provide a cytological definition of malignancy, as with astrocytomas and oligodendrogliomas a. WHO grade I subtypes i. meningothelial meningioma—exhibits groups of tumor cells surrounded by thin collagenous septa that are difficult to visualize, giving the impression of a syncytium ii. fibrous meningioma—appears similar to dense connective tissue iii. transitional meningioma—exhibits clumps of cells surrounded by whorls of more cells {onion bulbs}; has elements of meningothelial and fibrous subtypes iv. psammomatous meningioma—exhibits numerous laminated calcifications at the center of onion bulbs {psammoma bodies} that may become confluent and calcified v. angiomatous meningioma—appears similar to the meningothelial subtype but has a pronounced vascular component vi. secretory meningioma—focal epithelial differentiation forms a lumen containing accumulation of glucose polymers (i.e., periodic acid Schiff-positive) vii. lymphocyte-rich meningioma—exhibits chronic inflammatory infiltrates Figure 5–7 Types of meningiomas: meningothelial (A); fibrous (B); transitional, demonstrating onion bulbs (C); psammomatous, demonstrating psammoma bodies (D). (Courtesy of Dr. C. Yamada) b. WHO grade II subtypes Figure 5–8 Meningioma with dural tails. (From Fischbein NJ et al. Teaching Atlas of Brain Imaging. Stuttgart, Germany: Georg Thieme; 2000:205, Fig. D. Reprinted by permission.) i. clear cell meningioma—meningothelial-like but cells have clear, glycogen-rich cytoplasm ii. chordoid meningioma—exhibits cartilage-like myxoid extracellular matrix iii. atypical meningioma—exhibits increased mitotic activity, hypercellularity, and necrosis c. WHO grade III subtype i. anaplastic/malignant meningioma—exhibits high mitotic index (> 20 mitoses per high-powered field) 2. Tumor location: falx cerebri > olfactory groove, sphenoid ridges, parasellar region 3. Genetics: risk of meningioma formation is increased by prior irradiation; exhibits direct or progressive development of aggressive tumors, as with astrocytomas a. mutations in the neurofibromatosis 2 (NF-2) gene on chromosome 22, which encodes the merlin/schwannomin protein that acts as a tumor suppressor: promotes development of low-grade meningiomas as part of neurofibromatosis type 2 b. numerous loss of chromosomal heterogeneities promote development of malignant meningiomas 4. Diagnostic testing: neuroimaging demonstrates dural-based masses with tails (Fig. 5–8) b. edema is prominent only with the secretory, atypical, or anaplastic/malignant subtypes 5. Treatment: surgery; may add irradiation for atypical and anaplastic/malignant subtypes 6. Prognosis: 20% 20-year-recurrence rate following complete resection, considering all subtypes; however, up to 80% recurrence rate with anaplastic meningiomas Pediatric Pineal Region Tumors (In Order of Occurrence) 1. General histology: All subtypes develop from pinealocytes that have neuroendocrine potential a. pinealocyte-derived cells express the synaptic marker protein synaptophysin, neuron-specific enolase, tau protein, and serotonin 2. Pineocytoma a. specific histology: tumors show lobular aggregates of well-differentiated pinealocytes arranged as rosettes (Fig. 5–9) Figure 5–9 Pineocytoma rosettes. (From Keating R et al. Tumors of the Pediatric Central Nervous System. Stuttgart, Germany: Georg Thieme; 2001:315, Fig. 22–8. Reprinted by permission.) i. under electron microscopy, pinealocytes may exhibit annulate lamellae-like photoreceptor cells of the retina b. treatment: surgery c. prognosis: 85% 5-year survival 3. Pineoblastoma
Tumors of the Nervous System
I. Central Nervous System Tumors
A. Astrocytic Tumors
Box 5.1
 Histologically benign tumors with a good prognosis have a low MIB-1-labeled Ki-67 ratio, if not undetectable MIB-1 labeling
Histologically benign tumors with a good prognosis have a low MIB-1-labeled Ki-67 ratio, if not undetectable MIB-1 labeling
 Histologically benign tumors with a poor prognosis have a high MIB-1-labeled Ki-67 ratio
Histologically benign tumors with a poor prognosis have a high MIB-1-labeled Ki-67 ratio
Box 5.5
 Glioblastoma
Glioblastoma
 Medulloblastoma
Medulloblastoma
 Primitive neuroepidermal tumors
Primitive neuroepidermal tumors
 Ependymoma
Ependymoma
 Pineoblastoma
Pineoblastoma
 Germinoma
Germinoma
 Choroid plexus papilloma
Choroid plexus papilloma
B. Oligodendrogliomas
C. Gliomatosis Cerebri
D. Meningiomas
E. Pineal Tumors (Box 5.8)
Box 5.8
 Germinoma
Germinoma
 Astrocytoma
Astrocytoma
 Pineocytoma
Pineocytoma
 Teratoma
Teratoma
 Dermoid
Dermoid
 Pineoblastoma
Pineoblastoma
![]()
Stay updated, free articles. Join our Telegram channel






Full access? Get Clinical Tree


