and Aditya G. Shivane1
(1)
Cellular and Anatomical Pathology Level 4, Derriford Hospital, Plymouth, UK
Abstract
Many different types of primary tumour occur within the central nervous system, and it is also an important site for secondary spread of tumours from elsewhere in the body. Tumours are classified using the World Health Organisation system, which also assigns a biological grade indicating the degree of malignancy without treatment. This chapter describes the principles of tumour classification, the key pathological features of the majority of tumour subtypes and highlights the role of genetic findings in predicting biological behaviour and tumour classification.
Keywords
Brain tumourGeneticsChemosensitivityRadiosensitivityWHO gradeTumours of the central nervous system (CNS) represent around 3 % of all malignancies, but are the second most common type of tumour in children and young adults and population-based studies indicate an overall incidence of intracranial tumours of approximately 21/100,000 per year [1]. For the purposes of this chapter, we have included some tumours that occur within the cranial and spinal cavities that are not derived from CNS tissues, such as tumours of the meninges, pituitary gland and peripheral nerves.
6.1 Classification
Tumour classification is based on the presumed cell of origin and biological grade of malignancy, which is derived from the microscopic appearances. Most neuropathologists use the World Health Organisation classification system for CNS tumours [2] which has a biological grading system from I-IV, with I being very slow growing and unlikely to spread, to IV being rapidly growing and ability to spread extensively within the CNS (spread outside of the CNS is rare for most primary tumours of the CNS). It should be noted that these biological WHO grades relate to the natural behaviour of the tumour without treatment, so that some WHO Grade IV tumours, (e.g. medulloblastomas), can have a prolonged survival with current therapy. In addition, different genetic subtypes of tumours of the same WHO histological type and grade, vary in their prognosis, which is likely to be reflected in future versions of the WHO classification system.
Although tumours from elsewhere in the body may spread to the CNS, approximately two thirds of intracranial tumours are primary, the majority of which are either of astrocytic origin, meningiomas, pituitary tumours or peripheral nerve sheath tumours. The frequency of each type of tumour varies with age and gender, for example gliomas are more common in men and meningiomas more common in women, and certain tumours such as medulloblastomas, ependymomas, pilocytic astrocytomas, craniopharyngiomas and choroid plexus tumours are more common in childhood.
Tumours may present with a variety of neurological symptoms including signs of raised intracranial pressure (sometimes due to hydrocephalus, particularly in the case of posterior fossa tumours), focal neurological deficits and epilepsy. The aetiology of tumours of the CNS is unknown in the vast majority of cases. Recent data suggests an association between long-term mobile phone and some cases of ipsilateral schwannoma, but evidence for an association with other types of tumour is inconclusive [3, 4]. Tumours may occur as a delayed complication of radiotherapy (particularly meningeal tumours and gliomas), Epstein Barr virus infection may cause lymphoma and there are a number of rare inherited genetic syndromes that predispose to neurological tumours (see Table 6.1).
Table 6.1
Familial tumour syndromes involving the CNS
Syndrome | Gene, locus | CNS tumours |
|---|---|---|
Neurofibromatosis type 1 | NF1/neurofibromin, 17q11 | Neurofibromas, malignant nerve sheath tumours, astrocytic tumours, especially pilocytic astrocytoma of optic nerve |
Neurofibromatosis type 2 | NF2/merlin, 22q12 | Schwannoma, meningioma, ependymoma |
Tuberous sclerosis | TS1, 9q34 or TS2, 16p13 | Sub-ependymal giant cell astrocytoma |
von Hippel-Lindau disease | VHL, 3p25 | Haemangioblastoma |
Li-Fraumeni syndrome | TP53, 17p13 | Astrocytic tumours, primitive neuroectodermal tumours, medulloblastoma, choroid plexus tumours |
Gorlin syndrome (Naevoid basal cell carcinoma syndrome) | PTCH, 9q22 | Medulloblastoma |
Cowden syndrome | PTEN, 10q23 | Dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos disease) |
Turcot syndrome | APC, 5q21 (and others) | Medulloblastoma, glioblastoma |
Melanoma astrocytoma syndrome | p16, 9p | Astrocytic tumour |
Familial retinoblastoma | Retinoblastoma gene, 13q | Retinoblastoma, pineoblastoma |
Multiple endocrine neoplasia type 1 | MEN1, 11q13 | Pituitary adenoma |
Rhabdoid tumour predisposition syndrome | INI1, 22q11 | Atypical teratoid/rhabdoid tumours |
6.2 Sending Specimens to the Laboratory
It is important that specimens are sent to pathologists with adequate clinical information to allow accurate interpretation of the histological appearances (see Box 3.1). Information that a pathologist requires includes age, gender, family history (particularly of tumours), past medical history (including any treatment such as chemotherapy, radiotherapy and dexamethasone, or previous tumours), symptoms (including duration) and imaging findings (including location of tumour and pattern of contrast enhancement). If preoperative embolisation has been used to reduce bleeding during surgery e.g. for meningiomas, the pathologist must be informed as this can cause tumour necrosis and an increased mitotic rate [5], which can be confused with malignant change. In many cases it may be helpful to the neurosurgeon to have a rapid intraoperative diagnosis to confirm that the tissue is diagnostic and/or to give a provisional histological analysis to guide the extent of surgical resection that is appropriate (see Box 3.2). If an intraoperative diagnosis is planned it is important to warn the pathologist in advance so that laboratory staff can be prepared to deal with a specimen promptly. It is important to inform the laboratory if there is any potential that the patient may harbour a category 3 or 4 pathogen, so that additional precautions can be taken to prevent risk of infection to laboratory staff. A system should be agreed with a local laboratory on how intraoperative specimens are sent, but in general, the specimen should be sent immediately to the laboratory fresh, and should not be allowed to dry out in the process as this will hinder interpretation. Rapid intraoperative diagnoses are made by either freezing the tissue and then cutting ‘frozen sections’ from this onto a glass slide, or by squashing a small sample of tumour onto a glass slide and making a ‘smear preparation’. Tissue for ‘permanent’ histology is placed in formalin and may be sent to the laboratory separately. In an increasing number of cases fresh tissue may also be required to allow more extensive genetic testing to be undertaken on the tumour, so local laboratory advice should be taken on how tumours should be sent.
6.3 Gliomas
6.3.1 Astrocytic Tumours
These are the most common type of primary CNS tumour, histologically show a resemblance to astrocytes, and express glial fibrillary acidic protein. The different subtypes are listed in Table 6.2.
Table 6.2
Astrocytic tumours
Subependymal giant cell astrocytoma (WHO Grade I) |
Pilocytic astrocytoma (WHO Grade I) |
Pilomyxoid astrocytoma (WHO Grade II) |
Pleomorphic xanthoastrocytoma (WHO Grade II) |
Diffuse astrocytoma (fibrillary, protoplasmic and gemistocytic subtypes) (WHO Grade II) |
Anaplastic astrocytoma (WHO Grade III) |
Glioblastoma (including giant cell glioblastoma and gliosarcoma) (WHO Grade IV) |
The term ‘diffuse astrocytic tumours’ is often used to describe diffuse astrocytomas, anaplastic astrocytomas and glioblastomas, as these tumours tend to be highly infiltrative, so cannot be completely excised surgically. Diffuse astrocytomas (Fig. 6.1) can occur anywhere in the CNS, but predominantly within cerebral hemispheres of adults, and although most are slow growing, as is usually not possible to resect them completely, they recur over a number of years. Average survival is around 5–8 years, and in about half of cases, these tumours transform to high grade tumours, either anaplastic astrocytomas or glioblastomas (Fig. 6.2). Both of these latter types of tumour occur in the same regions as diffuse astrocytomas, but generally in older adults and they show contrast enhancement on imaging. Histologically, these high grade tumours are more cellular than diffuse astocytomas and have frequent mitoses. In addition, glioblastomas have areas of necrosis and show vascular proliferation. Although glioblastomas may arise from lower grade tumours (secondary glioblastomas), the majority occur de novo (primary glioblastomas). Average survival for anaplastic astrocytomas is 2–4 years, and for glioblastomas are around 1 year, although this depends on a number of factors including clinical condition (Karnofsky performance scale), age, treatment and genetic changes within the tumour (see below), with around 5 % patients with glioblastomas surviving 2 years. A list of features used to grade diffuse astrocytic tumours is given in Table 6.3.
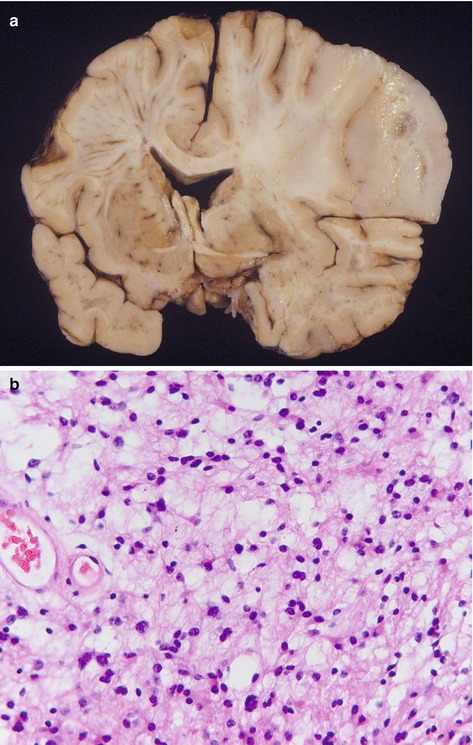
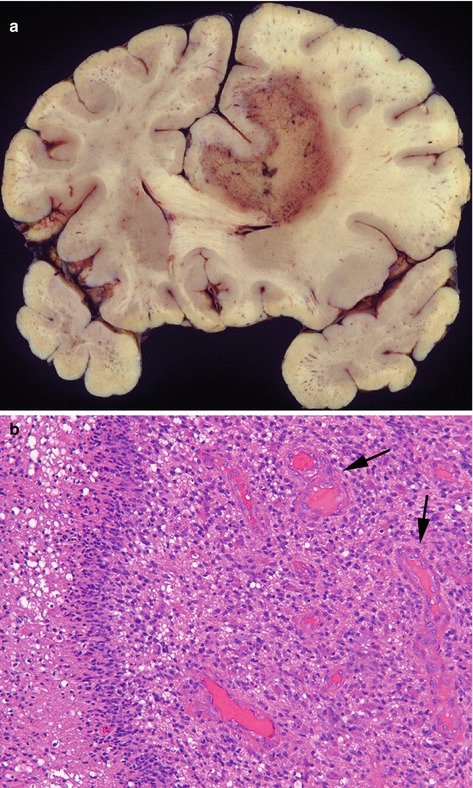
Fig. 6.1
(a) Diffuse astrocytoma, showing diffuse expansion of right upper cerebral hemisphere, shift of midline to the left, loss of grey/white matter demarcation and areas of cystic change. (b) Histologically is composed of cells with an astrocytic morphology with fairly uniform appearances, low cellularity and absence of mitotic figures. H&E stain
Fig. 6.2
(a) Glioblastoma predominantly in the white matter of the right frontal lobe with extensive necrosis (yellow area), causing shift of midline structures. (b) Microscopically the tumour is composed of astrocytic cells with large irregular nuclei, mitotic figures, area of necrosis (left) and vascular proliferation (arrows). H&E stain
Table 6.3
Histological features used in grading of diffuse astrocytic tumours
Increased cellularity – WHO Grade II |
Nuclear atypia – WHO Grade II |
Mitotic activity – WHO Grade III |
Vascular proliferation – WHO Grade IV |
Tumour necrosis – WHO Grade IV |
Gliomatosis cerebri is defined as a diffuse glioma (usually astrocytic, but sometimes oligodendroglial), involving at least three lobes, and often both cerebral hemispheres, brain stem and cerebellum. The extent of tumour can be seen on T2 and FLAIR weighted MRI, and although histological grading on biopsy can be difficult, most behave as WHO Grade III tumours.
Pilocytic astrocytomas are better circumscribed than the types of astrocytic tumour discussed above, only very rarely progress to more malignant tumours, are more common in children and young adults, and show a predilection for certain sites within the CNS (optic nerve, optic chiasm, hypothalamus and cerebellum). Histologically they are characterised by bipolar astrocytic cells forming solid and microcystic areas, Rosenthal fibres (hyaline accumulations of filamentous material), and vascular proliferation (Fig. 6.3). If they occur at a site where a surgical excision is possible, they have an excellent prognosis. Like high grade diffuse astrocytomas, these may show contrast enhancement on imaging. A related tumour, the pilomyxoid astrocytoma, most often occurring in the hypothalamic/chiasmic region of young children, has a more mucoid consistency and perivascular arrangement of tumour cells, but has tendency to recur and spread via the CSF.
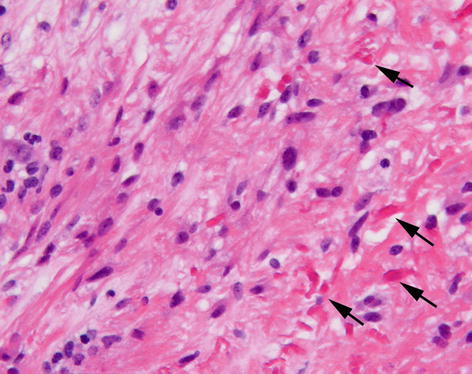
Fig. 6.3
Pilocytic astrocytoma composed of elongate astrocytic cells with irregular eosinophilic proteinaceous droplets (Rosenthal fibres – arrows). H&E stain
Pleomorphic xanthoastrocytomas are generally slow growing gliomas, but can occasionally be aggressive, and frequently present with seizures and on imaging have a cyst and mural nodule. The tumours often show quite marked cytological atypia and accumulation of cytoplasmic lipid, but usually lack mitotic activity.
Subependymal giant cell astrocytomas occur almost exclusively within the context of tuberous sclerosis and tend to form a predominantly exophytic mass protruding into the ventricles which may cause hydrocephalus. These tumours are composed of pleomorphic and multinucleate astrocytic cells, but are biologically benign.
6.3.2 Genetics of Gliomas
A large number of genetic changes have been described in gliomas, many of which are becoming of diagnostic and clinical relevance and are likely to feature prominently in future World Health Organisation classification systems. Some key genetic changes of clinical relevance are listed in Table 6.4.
Table 6.4
Clinically important genetic changes in gliomas
Genetic change | Comments |
|---|---|
Mutations in isocitrate dehydrogenase (IDH) genes types 1 and 2 | Present in about 75 % of diffuse astrocytomas, anaplastic astrocytomas, oligodendrogliomas and anaplastic oligodendrogliomas. Common in secondary glioblastomas. |
Common mutation in IDH1 gene (R132H) is responsible for about 90 % all mutations (Fig. 6.4). | |
Glioblastomas with IDH1 mutations have a better prognosis than those without. | |
Mutations in alpha-thalassaemia /mental retardation syndrome X–linked (ATRX) gene
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|





