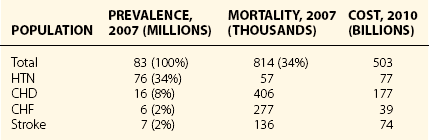
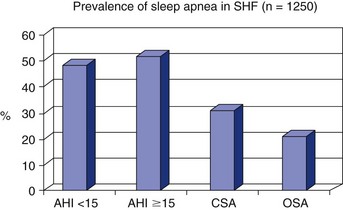
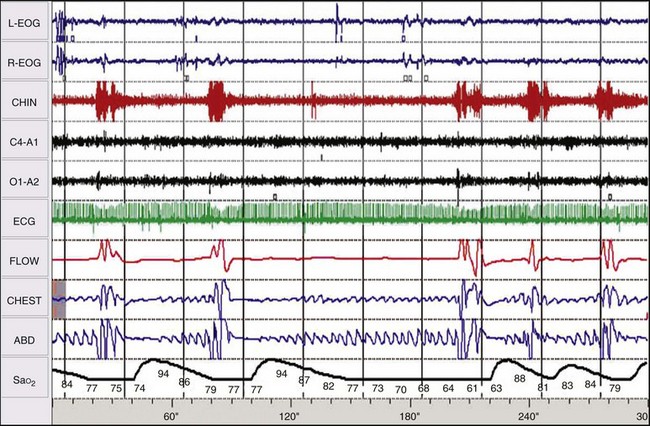
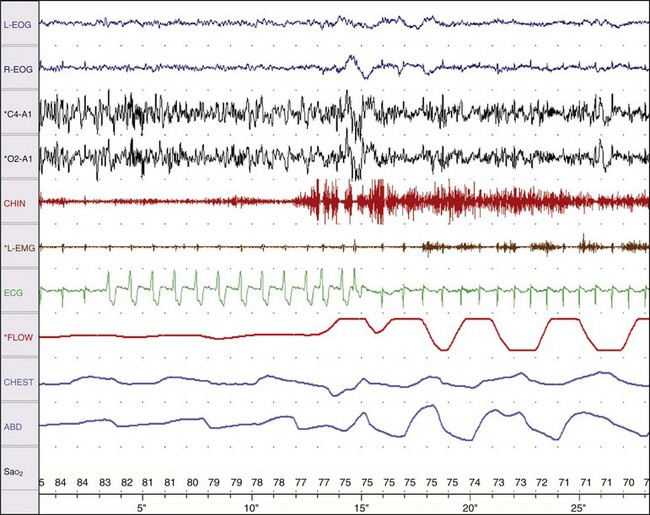
Chapter 14 14.1 Cardiovascular Diseases This chapter reviews cardiovascular diseases encountered in patients with sleep disorders. Both obstructive sleep apnea (OSA) and central sleep apnea (CSA) may be comorbid with a variety of cardiovascular diseases that continue to be highly prevalent and are associated with excess morbidity and mortality and huge economic costs. See Chapter 3.7 for a review of the regulation of cardiovascular physiology in sleep. An estimated 81 million Americans have one or more cardiovascular diseases. The prevalence of various cardiovascular diseases, the associated mortality, and the economic burden are presented in Table 14.1-1. Table 14.1-1 Prevalence, Mortality, and Economic Burden of Cardiovascular Disease in Adults in the United States CHD, chronic heart failure; CHF, congestive heart disease; HTN, hypertension. From Roger VL, Go AS, Lloyd-Jones DM, et al: Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation 2011;123:e18-e209. One of the most significant advances in clinical medicine has been our understanding that OSA and/or hypopnea, hitherto referred to collectively as OSA, may be either a cause of or may contribute to the progression, morbidity, and mortality of various cardiovascular diseases. OSA is characterized by recurrent upper airway occlusion that is either complete (apnea) or partial (hypopnea). The acute consequences of OSA include recurrent episodes of hypoxemia/reoxygenation; increases and decreases in Pco2; and large, negative swings in intrathoracic pressure. Figure 14.1-1 shows an example of OSA resulting in desaturation followed by reoxygenation with resumption of breathing. An arousal occurs at the termination of the apnea. In this patient, an esophageal balloon was inserted to measure intrathoracic pressure. Note the progressively increasing negative swings in intrathoracic pressure as a result of diaphragmatic contraction against the closed upper airway. The negative pressure swings are also reflected in the surrounding cardiac chambers, pulmonary vascular bed and interstitium, and the intrathoracic aorta, increasing the transmural pressure of the chambers and blood vessels. Increased transmural pressure of the atria may contribute to atrial premature excitation and other arrhythmias, such as atrial fibrillation. Increased left ventricular transmural pressure increases its afterload and oxygen consumption. Increased aortic transmural pressure increases the propensity for aortic dilation and dissection, whereas increased negative interstitial pressure increases the propensity for developing pulmonary edema. Figure 14.1-1 The three immediate nocturnal consequences of obstructive sleep apnea. Increasing evidence suggests that through these acute mechanisms, OSA leads to activation of redox-sensitive genes, oxidative stress, inflammatory processes, sympathetic overactivity, and hypercoagulability. All of these can contribute to endothelial dysfunction (Fig. 14.1-2), which is the underlying pathophysiologic mechanism that mediates a variety of cardiovascular disorders, including hypertension, atherosclerosis (coronary artery disease as well as cerebrovascular disease), pulmonary hypertension, right and left ventricular systolic and diastolic dysfunction, stroke, and even sudden death (Fig. 14.1-3). Figure 14.1-2 The consequences of recurrent sleep apneas and hypopneas include hypoxemia/reoxygenation in association with hypercapnia and hypocapnia, respectively, during apnea followed by hyperpnea. Figure 14.1-3 Proposed pathophysiologic pathways in the development of cardiovascular disease in obstructive sleep apnea. At times apneas can be very prolonged. This results in severe desaturation, as depicted in Figure 14.1-4. Disorders of cardiac rhythms can occur in patients with OSA. These dysrhythmias may include bradycardia, sinus pause (Fig. 14.1-5), heart block, ventricular ectopy and tachycardia (Fig. 14.1-6), and atrial fibrillation. Arrhythmias are apt to occur in patients with severe OSA and in those with cardiac disease. The mechanisms that mediate these dysrhythmias relate to the three major consequences of OSA: 1) altered blood gases that result in hypoxemia, hypercapnic acidosis, and hypocapnic alkalosis; 2) changes in autonomic tone; and 3) negative swings in intrathoracic pressure, which may distend the atria and ventricles. Particularly in the presence of coronary artery disease, the threshold for developing arrhythmias may be low. Figure 14.1-5 An example of sinus arrest in a patient with obstructive sleep apnea. (From Kapa S, Javaheri S: Constructive sleep apnea and arrhythmias. In Lee Chiong T, Javaheri S: Sleep medicine clinics: sleep and cardiovascular disease. Philadelphia, 2007, Elsevier, pp 575-581.) Through the mechanisms of endothelial dysfunction, which were reviewed earlier (see Figs. 14.1-2 and 14.1-3), OSA may either be a cause of or may contribute to coronary artery disease or its progression. Evidence is mounting, from both population and sleep clinic cohort studies, that OSA causes or contributes to the progression of coronary artery disease and eventually to elevated cardiovascular death and all-cause mortality. In a retrospective sleep laboratory cohort study, Peker and colleagues1 reported incident coronary artery disease in almost 25% of untreated OSA patients during a 7-year follow-up. The corresponding numbers in treated sleep apnea patients and nonapneic snorers were 4% and 6%, respectively. In an observational sleep laboratory cohort study of 1300 OSA patients and healthy study participants from Spain, Marin and colleagues2 observed that during a mean follow-up period of approximately 10 years, a 3 to 4 times higher incidence of fatal and nonfatal cardiovascular events was observed in the patients with severe OSA, defined as an apnea-hypopnea index (AHI) of 30 or more per hour, compared with simple snorers. Furthermore, in 372 patients with severe OSA who were compliant with CPAP, cardiovascular event rates were similar to those in nonapneic snorers. The Wisconsin Sleep Cohort Study3 is an 18-year mortality follow-up study (mean observation, approximately 14 years) conducted on a cohort sample of 1522 participants aged 30 to 60 years at baseline. The adjusted hazard ratio (HR) for all-cause mortality with severe versus no sleep-disordered breathing (SDB) was 3 (95% confidence interval [CI] = 1.4 to 6.3; Fig. 14.1-7). Importantly, after excluding the 126 participants who had used CPAP therapy, the association for all-cause mortality with severe versus no SDB became stronger, and the adjusted heart HR increased to 3.8 (CI = 1.6 to 9) for all-cause mortality and to 5.2 (CI = 1.4 to 19.2) for cardiovascular mortality (see Fig. 14.1-7). Figure 14.1-7 Probability of survival in patients with untreated obstructive sleep apnea. OSA is an independent risk factor for systemic arterial hypertension. Based on biologic plausibility (see Figs. 14.1-2 and 14.1-3), animal studies, longitudinal epidemiologic human studies that account for confounding factors, and therapeutic trials, evidence has evolved that indicates OSA is an important cause of hypertension. In the Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure, OSA is recognized as the first identifiable cause of hypertension.4 In a longitudinal study, Peppard and associates5 reported that the incidence of newly diagnosed arterial hypertension in a 4-year follow-up period increased as the AHI increased—a dose-dependent relationship. The increase in the risk for developing hypertension was doubled in those with an AHI of 5 to 15 events per hour, and a threefold increase in risk was seen with an AHI greater than 15 events per hour compared with study participants without a sleep disorder at baseline. From a clinical point of view, it is important to recognize that approximately half of the 70 million Americans who have systemic hypertension may have OSA. Furthermore, it is important to note that effective treatment of OSA has been shown to decrease systemic blood pressure. These reports are cited (Fig. 14.1-8) because of the power of the studies, the presence of a placebo control group that received sham CPAP, and the fact that the patients had hypertension to begin with. Collectively these studies show a small but significant decrease in hypertension in CPAP-treated patients (see Fig. 14.1-8), and hypertension improved the most in patients with the most severe sleep apnea and desaturation index and in those who used CPAP the most. Therefore it is safe to conclude that adequate therapy of OSA with CPAP is critical for reduction in blood pressure. Figure 14.1-8 Changes in hypertension induced by obstructive sleep apnea (OSA). Table 14.1-2 shows the prevalence of CSA and OSA in recent studies of patients with HFrEF. In the prevalence of these two forms of sleep apnea, reported variations have been considerable; this is due to a number of issues, such as the differentiation of obstructive from central hypopneas and the enrollment of obese patients, which increases the prevalence of OSA. Combining the results of the most recent series in the literature, as shown in Table 14.1-2, and using an AHI of 15 events per hour of sleep as the threshold, 31% of 1250 patients have CSA and 21% have OSA (Fig. 14.1-9). Table 14.1-2 Prevalence of Sleep Apnea in Recent Studies of Systolic Heart Failure AHI, apnea-hypopnea index; CSA, central sleep apnea; OSA, obstructive sleep apnea. Figure 14.1-9 Prevalence of sleep apnea in systolic heart failure (SHF). The largest study of patients with HFpEF involved 244 consecutive patients who were well characterized by right heart catheterization and echocardiography. Bitter and colleagues14 reported that 48% had an AHI of 15 or more events per hour, 25% with OSA and 23% with CSA (see Fig. 14.1-9). The hallmark of OSA in patients with HF is similar to that in patients with OSA in the general population, in that such patients are obese and have habitual snoring. In contrast, patients with CSA are normally thinner than patients with OSA, and the prevalence of habitual snoring is much less (Fig. 14.1-10). For these reasons, the suspicion for the presence of CSA in cardiology and in primary care clinics is low, so these patients often go undiagnosed. The risk factors for CSA in patients with HFrEF include a high New York Heart Association class, the presence of atrial fibrillation, more than 30 premature ventricular excitations per hour of sleep, the presence of one or more couplets per hour of sleep, the presence of one or more ventricular tachycardias per hour of sleep, a low steady-state awake Pco2 of less than 36 mm Hg, and a left ventricular ejection fraction of less than 20% (Fig. 14.1-11). Meanwhile, it is emphasized that in our studies6 the prevalence of excessive daytime sleepiness was similar in patients with systolic heart failure (SHF) with or without CSA and/or OSA (see Fig. 14.1-10). This further contributes to the underrecognition of sleep apnea in patients with SHF. Figure 14.1-10 Demographics, historical data, and physical examination findings in heart failure patients without sleep apnea, with central sleep apnea (CSA), and with obstructive sleep apnea (OSA).
Vascular Disorders
Impact of Cardiovascular Diseases
Effect of Sleep Disorders on Cardiovascular Physiology
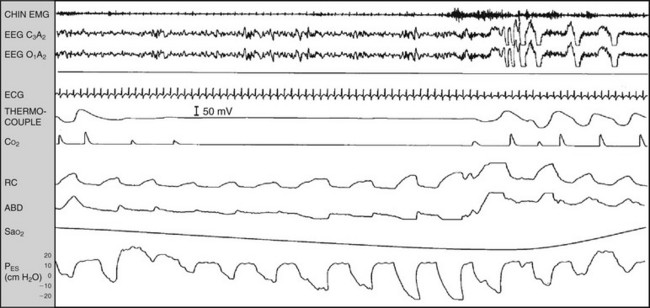
These include 1) altered blood gas chemistry, characterized by hypoxemia/deoxygenation and hypercapnia/hypocapnia; 2) arousals; and 3) large negative swings in intrathoracic pressure.
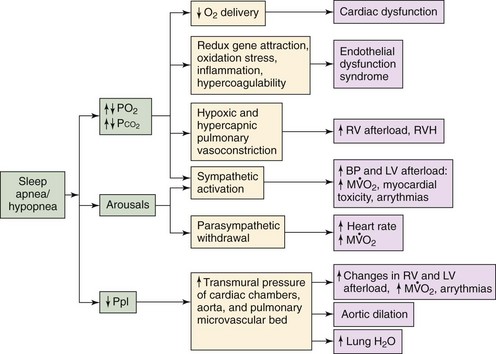
Other consequences are arousals and negative flows in pleural pressure, which in conjunction could eventually result in significant cardiovascular dysfunction. Hypoxemia may result in diminished oxygen delivery to the myocardium, which could result in arrhythmias and systolic and diastolic dysfunction. Hypoxemia/reoxygenation could result in activation of redox-sensitive genes, oxidative stress inflammation, and hypercoagulability leading to endothelial dysfunction syndrome. Both alveolar hypoxia and hypercapnia (as a result of sleep apnea/hypopnea) result in hypoxic and hypercapnic pulmonary vasoconstriction that increases right ventricular afterload and its myocardial oxygen consumption ( ). This eventually could result in right ventricular hypertrophy (RVH) and cor pulmonale. Hypoxemia and hypercapnia, along with arousals, could also lead to sympathetic activation that would increase blood pressure (BP). This would result in increased left ventricular afterload, increased
). This eventually could result in right ventricular hypertrophy (RVH) and cor pulmonale. Hypoxemia and hypercapnia, along with arousals, could also lead to sympathetic activation that would increase blood pressure (BP). This would result in increased left ventricular afterload, increased  , and arrhythmias. In addition, this increase in sympathetic activation and release of myocardial norepinephrine could result in myocardial toxicity and apoptosis. Arousals also result in parasympathetic withdrawal, which increases the heart rate and
, and arrhythmias. In addition, this increase in sympathetic activation and release of myocardial norepinephrine could result in myocardial toxicity and apoptosis. Arousals also result in parasympathetic withdrawal, which increases the heart rate and  . Finally, negative swings in pleural pressure (Ppl) increase the transmural pressure of the left ventricle (LV) and right ventricle (RV), both of which increase the wall tension of the ventricles, leading to an increase in
. Finally, negative swings in pleural pressure (Ppl) increase the transmural pressure of the left ventricle (LV) and right ventricle (RV), both of which increase the wall tension of the ventricles, leading to an increase in  . In addition, increasing the transmural pressure also affects the intrathoracic aorta and predisposes to aortic dilatation and potential dissection. Furthermore, increased pleural pressure of the atria predisposes to development of atrial fibrillation. Finally, a negative interstitial pressure increases transcapillary fluid flow, which facilitates the development of pulmonary edema. Overall, therefore, the consequences of sleep apnea and hypopnea in conjunction could result in cardiovascular dysfunction.
. In addition, increasing the transmural pressure also affects the intrathoracic aorta and predisposes to aortic dilatation and potential dissection. Furthermore, increased pleural pressure of the atria predisposes to development of atrial fibrillation. Finally, a negative interstitial pressure increases transcapillary fluid flow, which facilitates the development of pulmonary edema. Overall, therefore, the consequences of sleep apnea and hypopnea in conjunction could result in cardiovascular dysfunction.
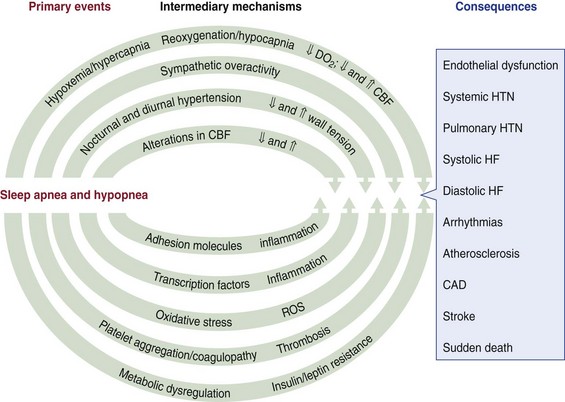
CAD, coronary artery disease; CBF, cerebral/coronary blood flow; DO2, dissolved oxygen; HF, heart failure; HTN, hypertension; ROS, reactive oxygen species.
Obstructive Sleep Apnea as a Cause of Cardiac Arrhythmias
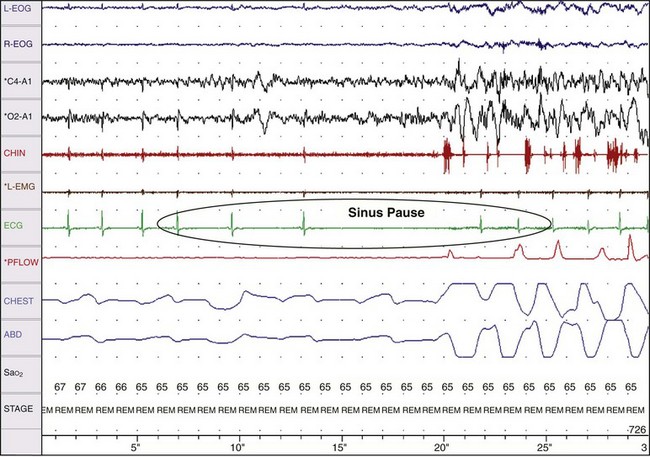
Obstructive Sleep Apnea as a Cause of Coronary Artery Disease
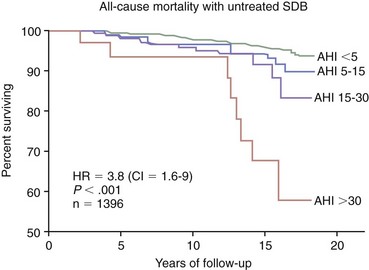
AHI, apnea-hypopnea index; CI, confidence interval; HR, hazard ratio; SDB, sleep-disordered breathing. (Modified from Young T, Finn L, Peppard PE, et al: Sleep-disordered breathing and mortality: eighteen-year follow-up of the Wisconsin Sleep Cohort. Sleep 2008;31:1071-1078.)
Obstructive Sleep Apnea as a Cause of Systemic Hypertension
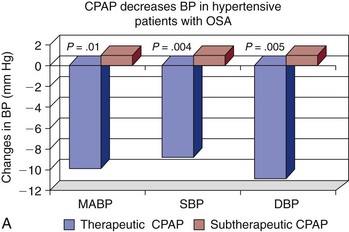
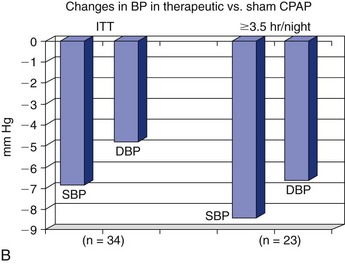
Note the improvement in hypertension when OSA is effectively treated with continuous positive airway pressure (CPAP) and in those patients who are adherent to therapy. BP, blood pressure; DBP, diastolic blood pressure; ITT, intention to treat; MABP, mean arterial blood pressure; SBP, systolic blood pressure. (A, Data from Becker HF, Javaheri S: Systemic and pulmonary arterial hypertension in obstructive sleep apnea. In Lee Chiong T, Javaheri S: Sleep medicine clinics: sleep and cardiovascular disease. Philadelphia, 2007, Elsevier, pp 549-557. B, Data from Coughlin SR, Mawdsley L, Mugarza JA, et al: Cardiovascular and metabolic effects of CPAP in obese males with OSA. Eur Respir J 2007;29:720-727.)
Epidemiology
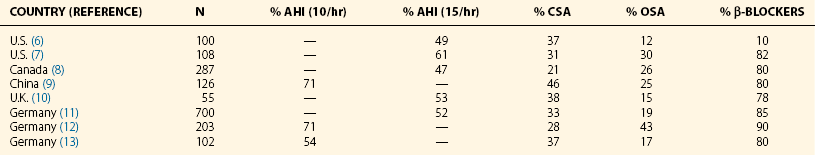
The data presented combine a series of world sleep studies (referenced in Table 14-2). AHI, apnea-hypopnea index; CSA, central sleep apnea; OSA, obstructive sleep apnea.
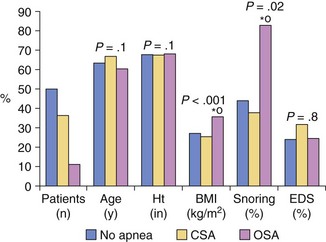
Patients with OSA were more obese and had a higher prevalence of habitual snoring than patients with CSA. No difference was found in prevalence of excessive daytime sleepiness between the patients with heart failure and the patients without sleep apnea. BMI, body mass index; EDS, excessive daytime sleepiness; Ht, height. (Modified from Javaheri S: Sleep disorders in systolic heart failure: a prospective study of 100 male patients. The final report. Int J Cardiol 2006;106:21-28.)![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Vascular Disorders
Only gold members can continue reading. Log In or Register to continue






