3
What Causes Epilepsy?
Introduction
There are many possible causes for epilepsy, including genetic, acquired, and provoking factors. Epilepsy can be classified into four categories based on presumed etiology:
The causes of epilepsy differ by age. In children and adolescents, epilepsy is more often genetically determined, whereas adult epilepsy is more often due to acquired structural causes. At every age, the cause of epilepsy is unknown for about half of individuals. Identification of the etiology of epilepsy has practical implication for both prognosis and treatment.
Idiopathic epilepsies
Idiopathic epilepsies are common, constituting about 40% of the epilepsies worldwide. They are characterized by generalized or partial seizures in otherwise normal infants, children, adolescents, and young adults with normal brain MRI and no previous relevant medical history. Response to antiepileptic drugs is usually satisfactory, but it is unproven that treatment changes ultimate outcome.
In most cases, idiopathic epilepsies exhibit a complex pattern of inheritance in which various genes act in a different way in each patient to produce the specific phenotype or syndrome. The various syndromes of idiopathic epilepsies differ in age of onset. Idiopathic epilepsies with complex (presumed polygenic) inheritance are divided into the idiopathic generalized epilepsies (IGEs) and the idiopathic partial epilepsies of childhood (Table 3.1).
Table 3.1. Idiopathic generalized and focal epilepsies.
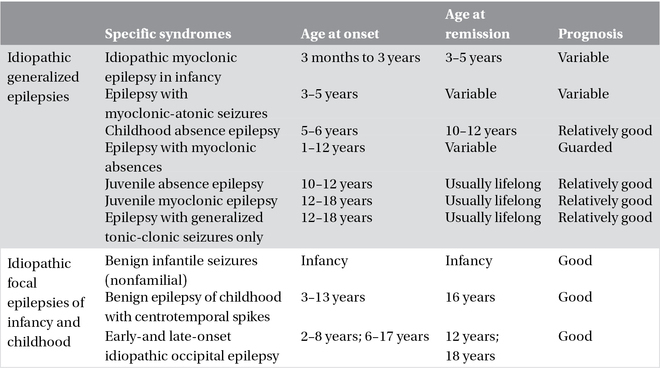
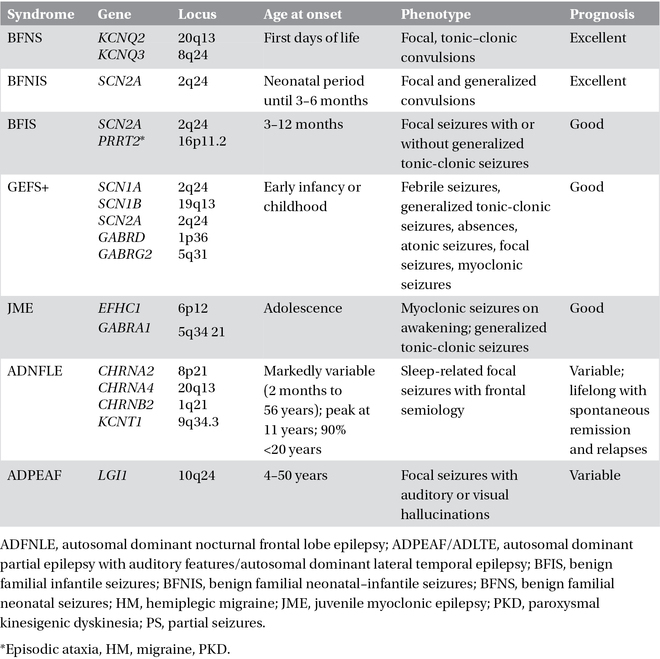
A few idiopathic epilepsy syndromes have a familial distribution with simple inheritance, usually autosomal dominant with reduced penetrance, and are usually caused by mutations in single genes that encode for ion channels or their accessory subunits. This has led to the concept that idiopathic epilepsies are channelopathies, even though the link between molecular deficit and clinical phenotype is still insufficiently characterized and non-ion channel genes are now emerging as causes of sporadic or familial early-onset focal, seemingly idiopathic seizure disorders. It is notable that mutations in the same gene can cause different epilepsy syndromes (phenotypic heterogeneity) and the same syndrome can be caused by mutations in different genes (genotypic heterogeneity). Phenotypic variability has been putatively attributed to modifier genes or polymorphisms determining the phenotypical expression or, alternatively, to environmental factors. Recent genetic insights have been gained through the discovery that monogenic idiopathic epilepsies may be comorbid with disorders such as paroxysmal movement disorders, hemiplegic migraine, broad-spectrum encephalopathies, learning difficulties, and psychiatric conditions. “Monogenic idiopathic epilepsies” are summarized in Table 3.2.
Symptomatic epilepsies
Symptomatic epilepsies are associated with structural brain abnormalities indicating an underlying disease or condition. This category includes (1) developmental and congenital disorders associated with genetic or acquired cerebral pathological changes, and also (2) acquired conditions. In symptomatic epilepsies, the underlying genetic conditions are responsible for either clear neuropathological abnormalities (e.g., epilepsy due to neurocutaneous diseases) or more subtle changes at the subcellular or molecular pathology level (e.g., the epilepsies due to Angelman syndrome, Rett syndrome, CDKL5 gene mutations).
Symptomatic epilepsies due to genetic or congenital disorders
Single-gene disorders
In most of the single-gene disorders that cause epilepsy and manifest primarily in childhood, seizures are only one symptom of a much broader clinical picture characterized by learning disabilities and other neurological features (Chapter 22). The seizures have variable characteristics and severity. These single-gene disorders are usually associated with variable and complex phenotypes. The epilepsy is distinctive, or is a predominant and consistent feature, in only a few of these conditions (Table 3.3).
Table 3.2. Monogenic idiopathic epilepsies.
Chromosomal disorders
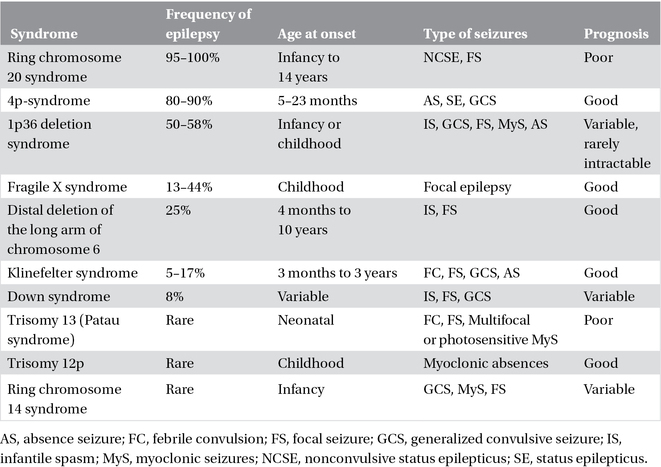
Epilepsy is frequently observed in chromosomal disorders (Chapter 22). These syndromes are associated with behavioral and intellectual disabilities, and characteristic dysmorphic features. In some, the clinical presentation and EEG abnormalities are characteristic (e.g., Angelman syndrome, ring chromosome 20 syndrome, and 4p-syndrome); in others, the manifestations appear nonspecific and are not diagnostic of the particular chromosomal abnormality. Often, seizure onset is during the neonatal period or in infancy (Table 3.4).
Inherited metabolic and mitochondrial disorders
Seizures are often part of the clinical picture of inherited metabolic and mitochondrial disorders, particularly when these conditions begin early in life (Chapter 23). Unfortunately, the clinical presentation of seizures is seldom distinctive enough to allow immediate diagnosis. Nevertheless, clinical phenotypes, epilepsy syndrome, and especially the characteristic times of presentation narrow diagnostic possibilities. EEG findings may also facilitate recognition of the more common diagnoses. All infants and young children seen with unexplained epilepsy and neurological disability (e.g., intellectual disability) should be evaluated for inherited metabolic disorder. A positive family history may provide an important clue, and careful studies of blood, urine, and cerebrospinal fluid (CSF) may reveal characteristic abnormalities. More common metabolic and mitochondrial diseases and associated epilepsy syndromes are reported in Table 3.5 and in Chapter 23.
Table 3.3. Complex single-gene disorders with epilepsy.
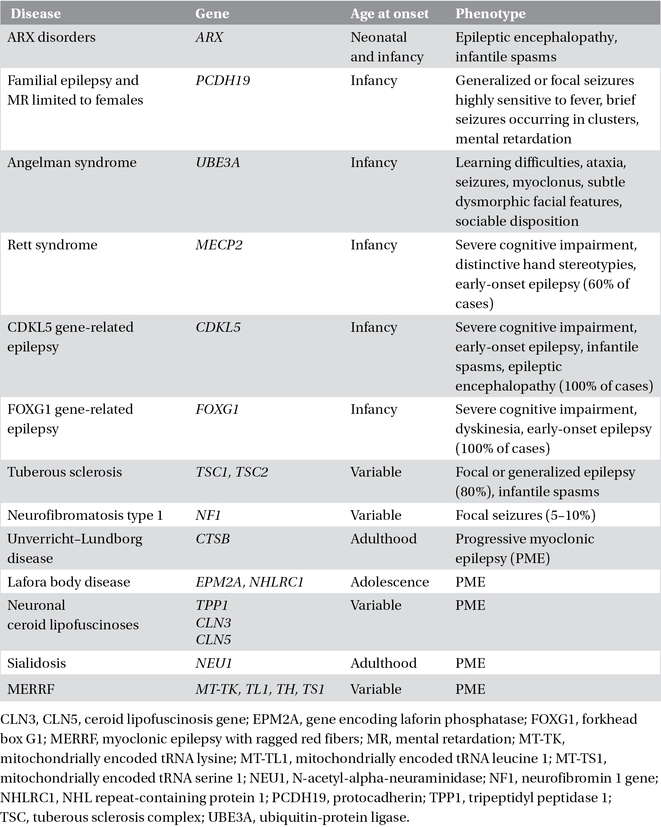
Table 3.4. Chromosomal disorders.
Malformations of cortical development
Malformations of cortical development include a wide range of disorders that commonly cause neurodevelopmental delay and epilepsy, especially in children. They account for at least 40% of drug-resistant childhood epilepsies. Abnormalities of neuronal and glial proliferation or programmed cell death, neuronal migration, synaptogenesis, and cortical organization result in distinct or partially overlapping entities. Some malformations are associated with mutations of specific genes, but others result from environmental influences such as intrauterine infection or perfusion failure. Although in many cases, the cause remains unclear, exome sequencing studies are revealing that de novo mutations of genes that encode for proteins regulating centrosome activity, vesicle trafficking, and cell adhesion are responsible for the majority of sporadic malformations. There is also emerging evidence that some regional malformations, such as hemimegalencephaly, are caused by somatic mosaic mutations that are present only in the malformed tissue.
The most common malformation of cortical development is focal cortical dysplasia (FCD). This term designates a spectrum of abnormalities of the laminar structure of the cortex, variably associated with cytopathological features including giant (or cytomegalic) neurons, dysmorphic neurons, and balloon cells. A developmental lineage model has been proposed in which balloon cells and dysplastic neurons are derived from radial progenitor cells in the telencephalic ventricular zone. The abnormal area is not usually sharply defined from the adjacent tissue. The close cytoarchitectural similarities between FCD and the cortical tubers of tuberous sclerosis has prompted the hypothesis of a common pathogenetic basis. Histopathologic similarities between FCD, hemimegalencephaly, and the dysembryoplastic neuroepithelial tumors, two highly epileptogenic developmental lesions, further support the hypothesis of a developmental origin.
A classification system with distinct clinicopathologic FCD variants has been proposed based on clinical presentation, imaging findings, and histopathologic features. FCD Type I refers to isolated lesions with either radial (FCD Type Ia) or tangential (FCD Type Ib) dyslamination of the neocortex, microscopically identified in one or multiple lobes. FCD Type II is an isolated lesion characterized by cortical dyslamination and dysmorphic neurons without (Type IIa) or with balloon cells (Type IIb). FCD Type III occurs in combination with hippocampal sclerosis (FCD Type IIIa) or with epilepsy-associated tumors (FCD Type IIIb). FCD Type IIIc is found adjacent to vascular malformations, whereas FCD Type IIId is associated with epileptogenic lesions acquired in early life (i.e., traumatic injury, ischemic injury, or encephalitis).
Table 3.5. Metabolic and mitochondrial disorders.
| Metabolic disorders in the newborn |
| Nonketotic hyperglycinemia |
| Pyridoxine dependency |
| Molybdenum cofactor deficiency |
| Sulfiteoxidase deficiency |
| Peroxisomal disorders (Zellweger syndrome, neonatal adrenoleukodystrophy, acyl-CoA oxidase deficiency) |
| Urea cycle disorders |
| Maple syrup urine disease |
| Organic acidurias |
| Fatty acid oxidation defects |
| Defects of mitochondrial energy metabolism (pyruvate dehydrogenase deficiency, pyruvate carboxylase deficiency, Leigh syndrome) |
| Disorders of carbohydrate metabolism (fructose-1,6-bisphosphatase deficiency, early-onset multiple carboxylase deficiency) |
| Metabolic disorders of early infancy |
| Lysosomal disorders (GM2 gangliosidoses, GM1 gangliosidosis type I and type II, Krabbe disease) |
| Disorders of vitamin metabolism (biotinidase deficiency, methylenetetrahydrofolate reductase deficiency, congenital folate malabsorption) |
| GLUT1 deficiency syndrome |
| Organic acidurias |
| Aminoacidurias (phenylketonuria, hyperphenylalaninemias, tyrosinemia type III) |
| Urea cycle disorders (arginase deficiency) |
| Disorders of GABA metabolism |
| Menkes disease |
| Metabolic disorders of late infancy |
| Metachromatic leukodystrophy |
| Deficiency of α-N-acetylgalactosaminidase |
| Mucopolysaccharidoses |
| Neuronal ceroid lipofuscinoses |
| Defects of mitochondrial energy metabolism (POLG disease) |
| Carbohydrate-deficient glycoprotein syndrome |
| Metabolic disorders of childhood and adolescence |
| Homocystinuria |
| Diabetes mellitus |
| Adrenoleukodystrophy |
| Lysosomal disorders (sialidosis type I and type II, Gaucher disease type III) |
| Neuronal ceroid lipofuscinosis |
| Lafora disease |
| Defects of mitochondrial energy metabolism (POLG mutations; MERRF; MELAS) |
| Dentatorubral-pallidoluysian atrophy |
| Unverricht–Lundborg progressive familial myoclonic epilepsy |
MELAS, mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke; MERRF, myoclonic epilepsy with ragged red fibers; POLG, polymerase g.
The most frequent clinical presentation of FCD is a child or adolescent developing intractable partial epilepsy. However, infantile spasms may be the first manifestation. FCD is a frequent cause of focal status epilepticus. It is the most common pathological substrate in epilepsy surgery series, reaching up to 40%. FCD is rarely a restricted process. Surface and intracranial EEG recordings reveal focal positive spike discharges or high-frequency discharges, and ablation of tissue exhibiting these electrographic patterns correlates with outcome. Although patients can be operated on without invasive intracranial recordings, the best results are obtained when resections are guided by intracranial EEG investigations (Table 3.6).
Neurocutaneous disorders
The neurocutaneous disorders are a group of distinct conditions characterized by congenital dysplastic abnormalities involving the skin and nervous system. Tuberous sclerosis complex (TSC) is a dominant disorder manifested primarily by abnormalities of the CNS, the skin, and the kidney. About 80% of TSC patients develop epilepsy with about two-thirds presenting with seizures before the age of 2 years, often with infantile spasms. Sturge–Weber syndrome (SWS) is a nonfamilial phakomatosis in which a venous angioma of the leptomeninges is accompanied by an ipsilateral nevus flammeus of the skin supplied by the trigeminal nerve. Seventy percent of patients with SWS develop seizures within the first year of life, and almost all have epilepsy before 4 years of age. Unilateral convulsive status epilepticus occurs in about 50%, leaving half with permanent hemiplegia. Another neurocutaneous disorder, neurofibromatosis, has only a 5–10% occurrence of epilepsy. Other, less frequent, neurocutaneous conditions include hypomelanosis of Ito, epidermal nevus syndrome, midline linear nevus syndrome, incontinentia pigmenti, and Klippel–Trenaunay–Weber syndrome.
Table 3.6. Malformations of cortical development.
| Abnormalities of gyration |
| Agyria–pachygyria subcortical band heterotopia spectrum (LIS1, TUBA1A, RELN, ARX, DCX genes) |
| Diffuse polymicrogyria (TUBB2B gene) |
| Multilobar polymicrogyria |
| Schizencephaly (EMX2 gene) |
| Minor gyral abnormalities |
| Heterotopia |
| Periventricular nodular heterotopia (FLNA gene) |
| Subcortical nodular heterotopia |
| Subcortical band heterotopia (DCX and LIS1 genes) |
| Other gross malformations |
| Megalencephaly and hemimegalencephaly |
| Agenesis of the corpus callosum |
| Anencephaly and holoprosencephaly |
| Microcephaly |
| Cortical dysgenesis associated with neoplasia |
| DNET |
| Ganglioglioma |
| Gangliocytoma |
| Other cortical dysplasias |
| Focal cortical dysplasia |
| Tuberous sclerosis (TSC1 and TSC2 genes) |
DNET, dysembryoplastic neuroepithelial tumor.
Symptomatic epilepsies due to acquired conditions
This category includes cerebral pathological processes as well as epilepsies due to external or environmental causes. Seizures can result from anything that injures the brain, with the types of injury depending on age. Seizures in children often are caused by birth traumas, infections such as meningitis, and congenital abnormalities. Nonaccidental brain injury, often under recognized acutely, is a relatively rare cause of symptomatic epilepsy in children. In the middle years, seizures are commonly caused by head injuries, infections, alcohol, stimulant drugs, and medication side effects. In the elderly, a higher proportion of seizures are caused by brain tumors and strokes.
Cerebral palsy
Cerebral palsy is associated with epilepsy in about 50% of quadriplegic and hemiplegic forms, and in about 26% of spastic diplegic and in dyskinetic forms. Infantile spasms are observed in more than 15% of patients. Epilepsy is usually early-onset, may be focal or generalized, and has a variable course. Only 13% achieve a remission of 2 years or more. Children with cerebral palsy and epilepsy function at lower levels in terms of cognitive skills and memory than children with the same form of cerebral palsy without epilepsy. Those with smaller lesions and severe epilepsy do less well than those with larger lesions without epilepsy.
Hippocampal sclerosis
The terms hippocampal sclerosis (HS), Ammon’s horn sclerosis, and mesial temporal sclerosis designate subtly different entities featuring gliosis and neuronal loss in temporal structures. There are several pathologic subtypes of HS that are associated with additional pathologic changes, in some cases outside the hippocampus (dual pathology). The typical MRI features of HS are reduced volume, increased signal intensity on T2-weighted images, and abnormal morphology of hippocampus. There is no sex or side preference; 48–56% of cases are bilateral in postmortem studies. In children with temporal lobe epilepsy, MRI detected HS in 21% of those with new-onset seizures and in 57% of those with refractory seizures. HS is the most common pathological finding in mesial temporal lobe epilepsy (MTLE), accounting for approximately 70% of patients undergoing surgery for drug-resistant focal seizures. The clinical features of MTLE include an early initial injury (typically complicated febrile convulsions, but also central nervous system infections or head injury) followed by a variable latent interval of around 7–10 years before onset of spontaneous seizures in late childhood or adolescence. Seizures are characterized by a typical rising epigastric aura, followed by behavioral arrest, and a slowly progressing unresponsiveness, with a motionless stare or oroalimentary automatisms.
Cerebrovascular diseases/stroke
Seizures are a frequent complication of a stroke and can even be the presenting symptom. Early seizures occur within 7 days of stroke symptoms onset; late seizures occur thereafter. The early, acutely symptomatic seizures can predict the later development of epilepsy. Pathophysiological mechanisms of early seizures differ between ischemic and hemorrhagic stroke and include sudden development of a space-occupying lesion with mass effect, focal ischemia, and blood breakdown products. Late seizures correlate with gliosis and development of meningocerebral scars.
Intracranial hemorrhage (ICH) is frequently the result of penetrating head trauma (see following text). Nontraumatic ICHs are more prominent in children and young adults and more common among men than women. Causes of nontraumatic ICH are aneurysms, vascular malformations (arteriovenous malformations, venous angiomas, cavernous angiomas, and telangiectasias), acute or chronic hypertension, amyloid angiopathy, venous thrombosis, primary and metastatic brain tumor, vasculitis, and bleeding disorders (hemophilia, sickle cell disease, thrombocytopenia, and leukemia). Vascular malformations may cause epilepsy even in the absence of overt bleeding; cavernous angiomas and arteriovenous malformations are the most epileptogenic forms. About 50–70% of seizures occur within the first 24 h of ICH, and 90% in the first 3 days, with the overall 30 day risk of seizures being about 8%. Factors predisposing to seizures include hemorrhagic size, cerebral cortex involvement, hydrocephalus, intracranial midline shift, low Glasgow Coma Scale, and severe neurological deficit. Continuous video–EEG monitoring is advised to recognize early electrographic seizures and nonconvulsive episodes, which represent an underestimated and frequent complication of ICH. Overall incidence after ICH is 4.2–20% for clinical seizures and around 30% for subclinical seizures. Frequency of status epilepticus ranges from 0.3% to 21.4%.
Epilepsy almost always develops within 2 years of the hemorrhage, and the risk of epilepsy is about 1%. In patients with an ischemic stroke, epilepsy occurs in about 6% of patients within 12 months and 11% within 5 years. Factors associated with a great risk of epilepsy are severity, greater size of infarct, hemorrhagic transformation, a cortical site of stroke, and embolism.
Related posts:
 Ketogenic Diet and Alternative Therapies
Using Parenteral Antiepileptic Medications
Recognizing Seizures and Epilepsy: Insights from Pathophysiology
Workup of New-Onset Seizures
When Should Vagus Nerve Stimulation Be Considered, and What Can It Accomplish?
Recognizing, Assessing, and Treating Seizures and Status Epilepticus in the ICU
Ketogenic Diet and Alternative Therapies
Using Parenteral Antiepileptic Medications
Recognizing Seizures and Epilepsy: Insights from Pathophysiology
Workup of New-Onset Seizures
When Should Vagus Nerve Stimulation Be Considered, and What Can It Accomplish?
Recognizing, Assessing, and Treating Seizures and Status Epilepticus in the ICU
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


