, Jean Paul G. Vonsattel2, Helmut Heinsen3, 4 and Horst-Werner Korf5
(1)
Dr. Senckenbergisches Chronomedizinisches Institut, Goethe University Frankfurt, Frankfurt, Germany
(2)
Medical Center Neurological Institute, Columbia University, New York, NY, USA
(3)
Division Psychiatic Clinic Morphological Brain Research Unit, Julius Maximilians University Würzburg, Würzburg, Germany
(4)
University of Sao Paulo Medical School, Sao Paulo, Brazil
(5)
Dr. Senckenbergisches Chronomedizinisches Institut, Goethe University, Frankfurt, Frankfurt, Germany
7.1 Systematic Pathoanatomical Investigations of the Brainstem in Huntington’s Disease (HD)
The involvement of the brainstem is still not among the established degenerative features of HD (Bruyn et al. 1979; Koeppen 1989; Lange and Aulich 1986; Oyanagi et al. 1989; Rüb et al. 2014a; Vonsattel and DiFiglia 1998; Vonsattel et al. 1985). Although some of the unexplained HD disease symptoms (e.g., oculomotor dysfunctions) also suggested degeneration of select brainstem nuclei, damage to the brainstem and its possible clinical relevance in HD has been controversially discussed for many decades (see Sects. 6.2, 6.5, and 6.6). Previous assumptions that select nuclei of the brainstem may also undergo neurodegeneration during HD (i.e., dopaminergic and GABAergic substantia nigra, auditory superior olive, lateral vestibular nucleus, precerebellar inferior olive) (Figs. 1.4, 7.1, 7.2, and 7.3) have not been revisited or taken into account by current researchers (see Chap. 1) (Bruyn et al. 1979; Bollen et al. 1986; Koeppen 1989; Kremer et al. 1992; Lange and Aulich 1986; Lange et al. 1976; Lasker and Zee 1997; Leigh et al. 1983, 1985; Oyanagi et al. 1989; Rüb et al. 2009, 2014a; Vonsattel 2008; Vonsattel and DiFiglia 1998; Vonsattel et al. 1985; Walker 2007a, b). These assumptions, however, were confirmed by recent systematic pathoanatomical investigations of serial brainstem sections stained for neuronal Nissl material (with Darrow red) and lipofuscin pigment (with aldehyde-fuchsin) of clinically diagnosed and genetically confirmed HD patients that suffered from chorea, cognitive decline, and personality changes and also showed disease signs possibly related to brainstem damage (i.e., broad-based gait, gait imbalance, dysphagia, slowed horizontal saccades, inability to start horizontal saccades without initial head thrust, slowed and saccadic smooth pursuits, falls) (Rüb et al. 2014a).
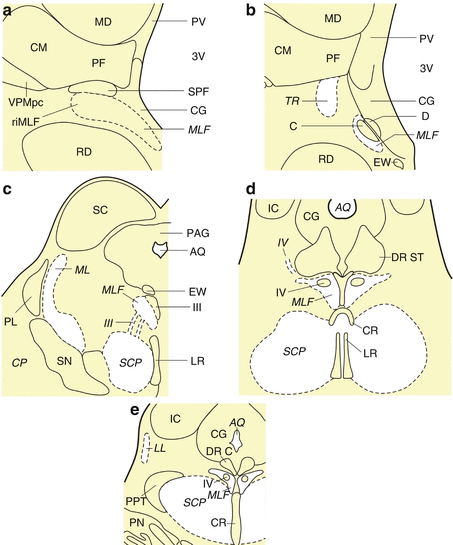
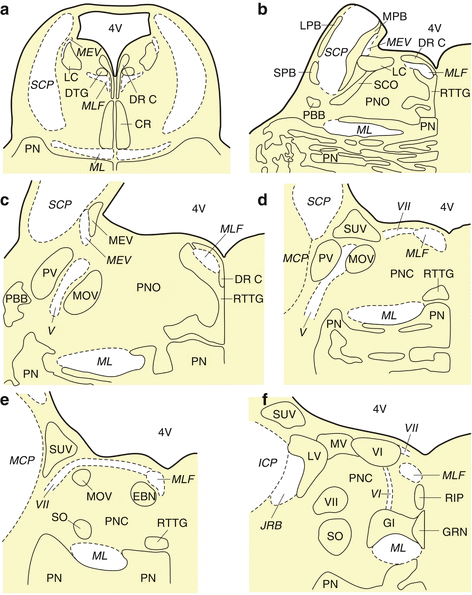
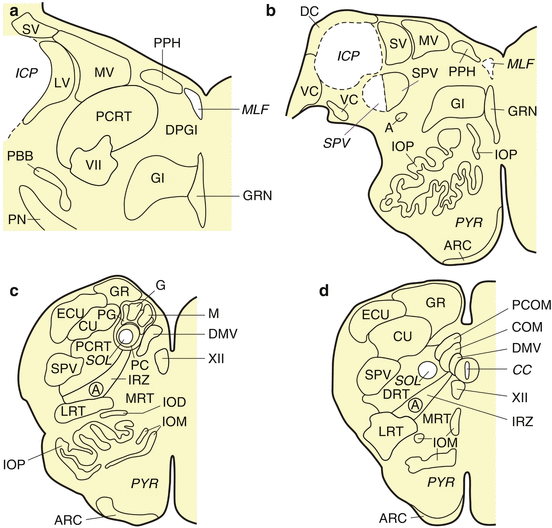
Fig. 7.1
The human midbrain. (a) Frontal section through the rostral midbrain at the level of the rostral interstitial nucleus of the medial longitudinal fascicle (riMLF). (b) Frontal section cut perpendicularly to the intercommissural axis of Forel showing the rostral midbrain at the level of the interstitial nucleus of Cajal (C). (c) Horizontal section through the caudal midbrain at the level of the dopaminergic substantia nigra (SN). (d) Horizontal section through the caudal midbrain at the level of the supratrochlear subnucleus of the serotonergic dorsal raphe nucleus (DR ST). (e) Horizontal section through the caudal midbrain at the level of the cholinergic pedunculopontine nucleus (PPT). Abbreviations: AQ aqueduct, CG central gray, CM centromedian nucleus of the thalamus, CR central raphe nucleus, CP cerebral peduncle, D nucleus of Darkschewitsch, DR C dorsal raphe nucleus, caudal subnucleus, DR ST dorsal raphe nucleus, supratrochlear subnucleus, EW Edinger-Westphal nucleus, C interstitial nucleus of Cajal, CR central raphe nucleus, IC Inferior colliculus, LL lateral lemniscus, LR linear raphe nucleus, MD mediodorsal nucleus of the thalamus, ML medial lemniscus, MLF medial longitudinal fascicle, PAG periaqueductal gray, PF parafascicular nucleus of the thalamus, PL paralemniscal nucleus, PN pontine nuclei, PV paraventricular nucleus of the thalamus, R red nucleus, riMLF rostral interstitial nucleus of the medial longitudinal fascicle, SC superior colliculus, SCP superior cerebellar peduncle, SN substantia nigra, SPF subparafascicular nucleus of the thalamus, TR tractus retroflexus (habenulo-interpeduncular tract), VPM ventral posterior medial nucleus of the thalamus, VPMpc ventral posterior medial nucleus of the thalamus, parvocellular part, III oculomotor nucleus, III oculomotor nerve, 3V third ventricle, IV trochlear nucleus, IV trochlear nerve
Fig. 7.2
The human pons. (a) Horizontal section through the rostral pons at the level of the central raphe nucleus (CR). (b) Horizontal section through the rostral pons at the level of the parabrachial nuclei (i.e., MPB medial parabrachial nucleus, LPB lateral parabrachial nucleus, SPB subpeduncular pigmented nucleus). (c) Horizontal section through the mid pons at the level of the trigeminal nuclei (i.e., MOV motor trigeminal nucleus, PV principal trigeminal nucleus, MEV mesencephalic trigeminal nucleus). (d) Horizontal section through the caudal pons at the level of the superior vestibular nucleus (SUV). (e) Horizontal section through the caudal pons at the level of the area of the excitatory burst neurons for horizontal saccades (EBN). (f) Horizontal section through the pontomedullary junction at the level of the raphe interpositus nucleus (RIP). Abbreviations: CR central raphe nucleus, DR C dorsal raphe nucleus, caudal part, DTG dorsal tegmental nucleus (Gudden), EBN area of the excitatory burst neurons for horizontal saccades, GI gigantocellular reticular nucleus, GRN great raphe nucleus, ICP inferior cerebellar peduncle, JRB juxtarestiform body, LC locus coeruleus, LPB lateral parabrachial nucleus, LV lateral vestibular nucleus, MCP medial cerebellar peduncle, MEV mesencephalic trigeminal nucleus, MEV mesencephalic trigeminal tract, ML medial lemniscus, MLF medial longitudinal fascicle, MPB medial parabrachial nucleus, MOV motor trigeminal nucleus, MV medial vestibular nucleus, PBB pontobulbar body, PN pontine nuclei, PNC pontine reticular formation, caudal nucleus, PNO pontine reticular formation, oral nucleus, PV principal trigeminal nucleus, RIP raphe interpositus nucleus, RTTG reticulotegmental nucleus of the pons (nucleus Bechterew), SCO subcoeruleus nucleus, SCP superior cerebellar peduncle, SO superior olive, SPB subpeduncular pigmented nucleus, SUV superior vestibular nucleus, 4V fourth ventricle, V trigeminal nerve, VI abducens nucleus, VI abducens nerve, VII facial nucleus, VII facial nerve
Fig. 7.3
The human medulla oblongata. (a) Horizontal section through the caudal pontomedullary junction at the level of the caudal pontobulbar body (PBB). (b) Horizontal section through the caudal pontomedullary junction with the dorsal (DC) and ventral cochlear (VC) nuclei. (c) Horizontal section through the rostral medulla oblongata with the principal nucleus of the inferior olive (IOP). (d) Horizontal section through the caudal medulla oblongata with the central canal (CC). Abbreviations: A ambiguus nucleus, ARC arcuate nucleus, COM commissural solitary nucleus, CU cuneate nucleus, DC dorsal cochlear nucleus, DMV dorsal motor vagal nucleus, DPGI dorsal paragigantocellular reticular nucleus, DRT dorsal reticular nucleus, ECU external cuneate nucleus, G gelatinous solitary nucleus, GR gracile nucleus, GRN great raphe nucleus, ICP inferior cerebellar peduncle, IOD inferior olive, dorsal subnucleus, IOM inferior olive, medial subnucleus, IOP inferior olive, principal subnucleus, IRZ intermediate reticular zone, LRT lateral reticular nucleus, LV lateral vestibular nucleus, M medial solitary nucleus, MLF medial longitudinal fascicle, MRT medial reticular nucleus, MV medial vestibular nucleus, PBB pontobulbar body, PC parvocellular solitary nucleus, PCOM paracommissural solitary nucleus, PCRT parvocellular reticular nucleus, PG pigmented solitary nucleus, PN pontine nuclei, PPH prepositus hypoglossal nucleus, PYR pyramidal tract, SOL solitary tract, SPV spinal trigeminal nucleus, SPV spinal trigeminal tract, SV spinal vestibular nucleus, VC ventral cochlear nucleus, 4V fourth ventricle, VII facial nucleus, XII hypoglossal nucleus
The pathoanatomical approach with unconventional thick serial brain tissue sections complements the routine neuropathological examination of single thin brain tissue sections and allows the recognition of subtle pathological changes and reveals more detailed information with respect to the extent and severity of the pathological processes of neurodegenerative diseases than the routine neuropathological approach (Figs. 5.2, 5.3, 5.4, 5.5, 6.4, and 6.5). This pathoanatomical approach was applied in recent systematic HD brainstem studies and disclosed a consistent and widespread degeneration of a variety of brainstem nuclei in HD patients (Rüb et al. 2008b, 2009, 2014a) (Figs. 6.4 and 6.5). Within the framework of these systematic studies, serial thick brainstem sections were also immunolabeled with an antibody directed against the protein aggregation marker p62 to identify neuronal aggregations of the disease protein of HD. The degradation-related, multifunctional, proteasomal shuttle and ubiquitin-interacting protein p62 together with ubiquitin participates in the intracellular trafficking, removal, and degradation of short-lived or aberrantly structured proteins. It serves the presentation of polyubiquitinated protein substrates to the 26S proteasome and interacts with polyubiquitin-tagged protein substrates, as well as with the proteolytic proteasome. The ubiquitin-association domain (UBA) of p62 binds covalently to polyubiquitin chains and enables low-affinity interaction with them. The ubiquitin-like domain (UbL) in the N-terminus of p62 binds to and directly interacts with the multimeric 26S proteasome. p62 is also known to be a structural component of the abnormal nuclear and/or cytoplasmic neuronal aggregations of the polyubiquitinated disease proteins in various human neurodegenerative diseases (see Sects. 8.2 and 8.3) (Figs. 8.3, 8.4, and 8.5) (Kuusisto et al. 2001, 2008; Ortega et al. 2007; Rüb et al. 2014a; Seibenhener et al. 2004; Seidel et al. 2009, 2010; Wooten et al. 2006).
Double immunostaining procedures were performed to identify and examine the topographical and intra-axonal localization, as well as composition of p62 immunoreactive aggregates in the brainstem neuropil. The AT270 antibody, directed against the neuronal cytoskeletal protein tau, was used as axonal marker, and the anti-p62 antibody served as marker for axonal protein aggregates (Figs. 9.2, 9.3, 9.4, and 9.5) (Goedert et al. 1994; Rüb et al. 2014a; Shaw et al. 1981). In addition, the 1C2 antibody was used to confirm the presence of huntingtin (Htt) with an expanded polyglutamine sequence of more than 37 repeats in the axonal aggregates. An anti-ubiquitin antibody was applied to investigate the additional association of axonal aggregates with the small degradation-related 8 kDa protein ubiquitin assigned to the family of the heat shock proteins (HSP) (Figs. 9.4 and 9.5) (Rüb et al. 2014a; Seidel et al. 2010; Trottier et al. 1995). The immunocytochemical findings of these recent systematic brainstem studies are detailed in Sect. 9.2.
7.2 Neurodegenerative Brainstem Features in Huntington’s Disease (HD)
The brainstem of HD patients (Vonsattel grades 2–4 of striatal atrophy) showed no macroscopical signs of degeneration except for a slightly flattened pons (Fig. 7.4) (Rüb et al. 2014a). Light microscopical examination of thick brainstem tissue sections, however, showed that brainstem affection in HD is more widespread than believed so far. It revealed a consistent neuronal loss in the dopaminergic compact part and GABAergic reticulate part of the substantia nigra, in the precerebellar pontine nuclei and inferior olive, as well as in the oculomotor reticulotegmental nucleus of the pons, in the premotor oculomotor area of the excitatory burst neurons for horizontal saccades and raphe interpositus nucleus, in the auditory superior olive, as well as in the lateral and medial vestibular nuclei. In some of the HD patients studied, a mild neuronal loss was also present in the superior vestibular nucleus, as well as in the ingestion-related facial and parvocellular reticular nuclei (Figs. 5.7, 6.2, 6.3, 6.4, 6.5, 7.2, and 7.3).
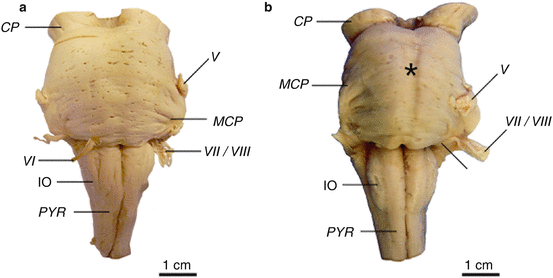
Fig. 7.4
The lower brainstem in Huntington’s disease (HD). (a) Ventral aspect of the lower brainstem of a typical control individual. (b) The lower brainstem with a flattened pons (asterisk) of a clinically diagnosed and genetically confirmed Huntington’s disease (HD) patient (Vonsattel grade 3 of neostriatal atrophy) (Reprinted from Rüb et al. (2014a), (Figure 1, page 251); with kind permission from John Wiley and Sons). Abbreviations: CP cerebral peduncle, IO inferior olive, MCP medial cerebellar peduncle, PYR pyramidal tract, V trigeminal nerve, VI abducens nerve, VII facial nerve, VIII vestibulocochlear nerve
Neuronal loss in the brainstem nuclei did not correlate with the length of the CAG repeats in the mutated HD allele, age at HD onset, duration of HD, age at death, or the Vonsattel grade of striatal atrophy. GFAP immunoreactive astrocytes were present in all degenerated and spared brainstem nuclei of all HD patients (Rüb et al. 2014a).
7.3 Clinical Relevance of Brainstem Neurodegeneration in Huntington’s Disease (HD)
Although its involvement is not yet among the currently established degenerative features of HD, damage to the brainstem underpins that neurodegeneration during HD is not restricted to the neostriatum and select areas of the cerebral neo- and allocortex. It provides additional support for the currently favored opinion that HD is a multisystem neurodegenerative disease, which is characterized by a distribution pattern of brain damage that shows more overlap with that of the related polyglutamine ataxias (e.g., spinocerebellar ataxias types 1, 2, and 3) than has been believed so far (Atkin and Paulson 2014; Borrell-Pagès et al. 2006; Braak and Braak 1992a, b; Bruyn et al. 1979; De la Monte et al. 1988; Estrada-Sanchez and Rebec 2013; Fennema-Notestine et al. 2004; Ferrante et al. 1987; Finkbeiner and Mitra 2008; Hedreen et al. 1991; Heinsen et al. 1992, 1994, 1996, 1999; Heinsen and Rüb 1997; Imarisio et al. 2008; Lange 1981; Lange and Aulich 1986; Lange et al. 1976; Lastres-Becker et al. 2008; Li and Conforti 2013; Margolis and Ross 2003; Myers et al. 1988; Riess et al. 2008; Rüb et al. 2003a, 2004a, b, 2005, 2008a, 2009, 2013b; Schulte and Littleton 2011; Seidel et al. 2012; Selemon et al. 2004; Sieradzan and Mann 2001; Sotrel et al. 1991; Valera et al. 2005; Vonsattel 2008; Vonsattel and DiFiglia 1998; Vonsattel et al. 1985; Walker 2007a,b). As degeneration of the brainstem nuclei did not correlate with the Vonsattel HD grade of neostriatal atrophy, it is likely that damage to these subcortical nuclei does not develop simultaneously with, but independently from the well-known pathognomonic lesions in the neostriatum (Rüb et al. 2014a).
Related posts:
 Pathological Nerve Cell Alterations in Huntington’s Disease (HD) and Their Possible Role for the Demise of Nerve Cells
Conclusions and Outlook
Pathological Nerve Cell Alterations in Huntington’s Disease (HD) and Their Possible Role for the Demise of Nerve Cells
Conclusions and Outlook
 Elucidation of the Role of the Premotor Oculomotor Brainstem Nuclei in the Pathogenesis of Oculomotor Dysfunctions in Huntington’s Disease (HD)
Elucidation of the Role of the Premotor Oculomotor Brainstem Nuclei in the Pathogenesis of Oculomotor Dysfunctions in Huntington’s Disease (HD)
 Intraneuronal Transport and Defense Mechanisms with Possible Pathogenetic Relevance in Huntington’s Disease (HD)
Intraneuronal Transport and Defense Mechanisms with Possible Pathogenetic Relevance in Huntington’s Disease (HD)
 Degeneration of Select Motor and Limbic Nuclei of the Thalamus in Huntington’s Disease (HD)
Degeneration of Select Motor and Limbic Nuclei of the Thalamus in Huntington’s Disease (HD)
 Consistent and Widespread Degeneration of the Cerebellum in Huntington’s Disease (HD)
Consistent and Widespread Degeneration of the Cerebellum in Huntington’s Disease (HD)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


