 TIPS AND TRICKS
TIPS AND TRICKSAtypical presentations of common diseases can complicate and delay the diagnosis of the patient with YOD. At the same time, multiple pathologies share overlapping phenotypes. Individuals with YOD are also more likely to be misdiagnosed as having a primary psychiatric disease. Due to a combination of factors, the duration from onset of symptoms to referral is significantly longer than that for persons with late onset dementia (LOD). A wide range of pathologies exist for the underlying cause of YOD, including neurodegenerative disorders, vascular diseases, autoimmune and inflammatory diseases, infections, toxin exposure, and adult onset mitochondrial, storage, and metabolic disorders. As in LOD, neurodegenerative disorders account for the etiology in the majority of patients with EOD; however, young individuals with dementia are also more likely than elderly individuals to have a familial or non-degenerative cause.
Approach to diagnosis
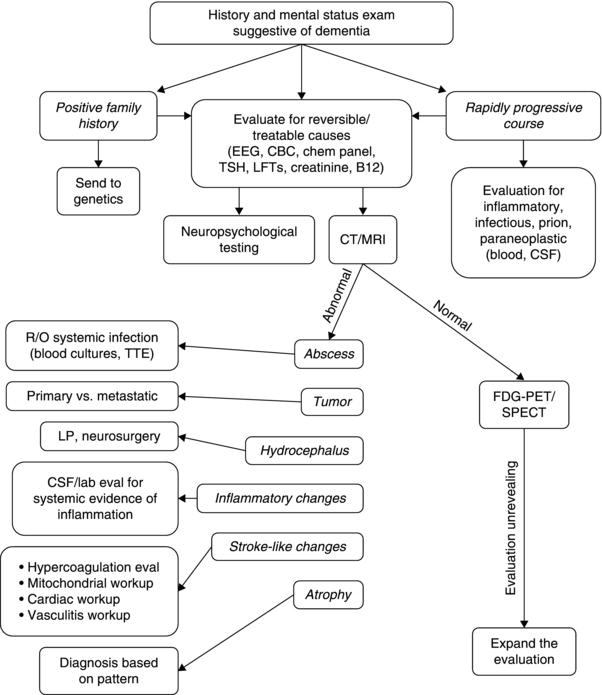
The etiology of dementia in a young individual should be pursued using a standardized clinical approach (Figure 3.1). The clinical evaluation begins with obtaining a detailed history from the patient and a reliable informant regarding the nature of cognitive and behavioral symptoms, rate of decline, and evolution of symptoms over time. A thorough inquiry of any accompanying medical or neurological symptoms should be included. A review of the patient’s medical history should include any history of malignancy, autoimmune disease, diabetes mellitus, cardiomyopathy, or conduction defect. The patient should be questioned about history of head injury, seizures, hearing loss, visual impairment including cataracts, weight loss, or change in bowel habits. History of tobacco use, illicit drug or alcohol use, travel, and exposure to heavy metals, solvents or inhalants should be addressed. It is important to review performance in school, history of a learning disability, educational attainment, and occupation.
Figure 3.1 Recommendations for a staged approach to the evaluation of a patient with young onset dementia.
 CAUTION
CAUTIONA family history of a dementia syndrome and/or other neurological symptoms should be aggressively explored in all patients with YOD, keeping in mind that phenotypes of a disorder may be variable within a family. A complete family history should be collected, paying attention to members with cognitive impairment or dementia, parkinsonism, motor neuron disease, mental retardation or developmental delay, stroke, death at young age, seizure disorder, psychiatric disease, autoimmune disease, or malignancy. At times a negative family history can be misleading if a family member with the mutation in question has died before the onset of neurological symptoms or in the case of a sporadic mutation. Table 3.1 provides a summary of some familial disorders that can present with YOD as well as possible co-existing neurological and medical symptoms and signs.
During the diagnostic evaluation, the physician should meet with the patient and family regularly to readdress their goals and reassess how extensive an evaluation they would like to pursue. Families should be aware that arriving at a diagnosis may provide information for the offspring of a patient with YOD in whom a genetic disorder is highly suspected and could, therefore, influence their decision about future children.
Diagnostic testing
Round 1
The rapidity of symptom onset can help to narrow the differential diagnosis. Precipitous decline in cognition often indicates an underlying vascular or infectious process affecting the central nervous system, or an acute toxic or metabolic insult. Initial laboratory studies must include a complete blood count, electrolytes, blood urea nitrogen, creatinine, liver function tests, ammonia, thyroid function tests, and toxicology screen. Emergency evaluation with neuroimaging to exclude an ischemic infarct, intracranial hemorrhage, meningitis, encephalitis, or vasculitis is mandatory. If neuroimaging and laboratory studies do not identify a cause for the acute change, a lumbar puncture to evaluate for evidence of infection or inflammation is necessary and empiric treatment with antimicrobials must be seriously considered based on the clinical circumstance. If initial lumbar puncture results (i.e. cell count, protein, and glucose) do not suggest the presence of a central nervous system (CNS) infection, an electroencephalogram (EEG) ought to be performed to rule out non-convulsive status epilepticus. If this work-up is unrevealing, then frequently neuroimaging, cerebrospinal (CSF) exam, and/or EEG may need to be repeated. Prolonged EEG monitoring may also be considered.
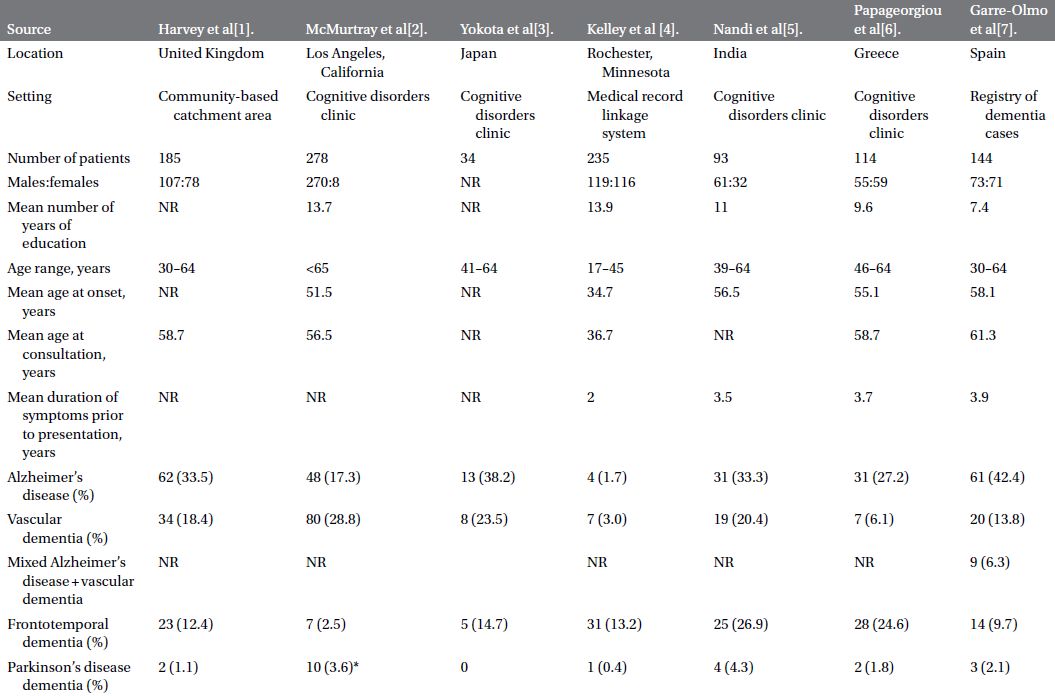
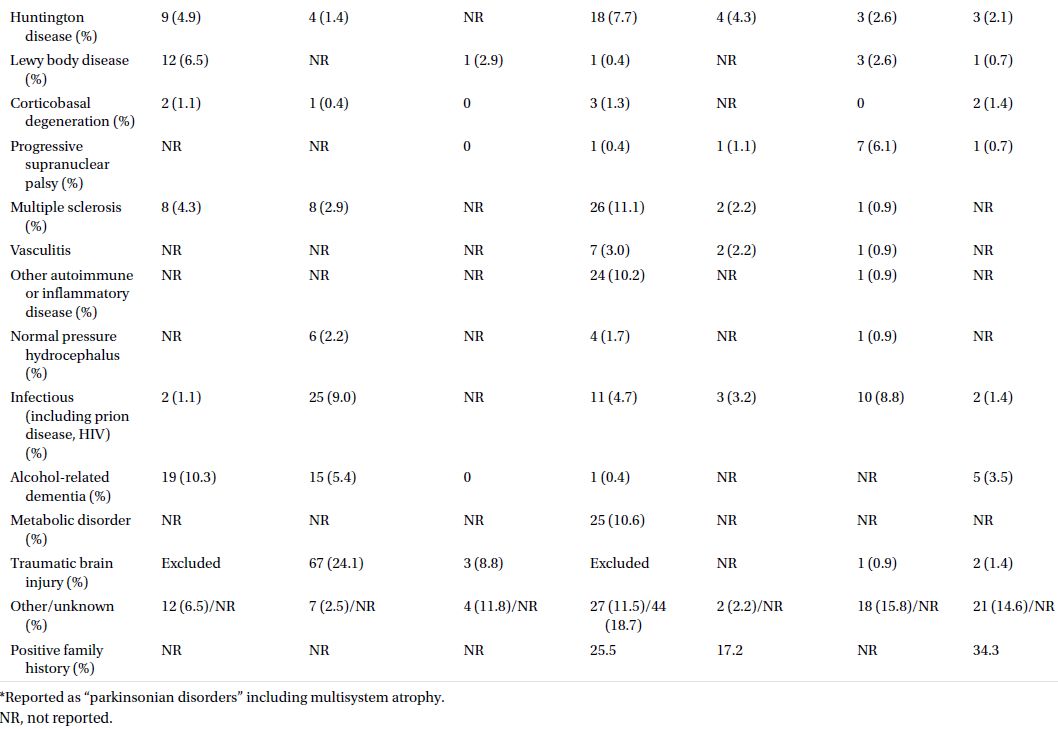
An insidious rate of cognitive and functional decline has a much broader differential. Epidemiological studies on cohorts of patients with YOD have identified a variety of pathological substrates (Table 3.2). A comprehensive and meticulous general and neurological examination can guide the physician in the proper direction. Special attention should be given to the presence or absence of oculomotor abnormalities, pyramidal signs, parkinsonism, dystonia, chorea, myoclonus, cerebellar signs, peripheral neuropathy, or fasciculations.
Diagnostic studies are performed to identify neurodegenerative and non-neurodegenerative causes of cognitive decline, often with the hope that a treatable etiology will be identified. The evaluation of the patient with a dementia syndrome should proceed in a thoughtful, stepwise manner in order to be efficient, conscientious of costs, and thorough while keeping in mind the wishes of the patient. The initial investigations should evaluate for treatable etiologies followed by a focused approach based on the differential generated after clinical history and examination. There are times when the initial studies do not identify the underlying etiology but the abnormalities serve to narrow the differential. If these attempts are fruitless then the differential must expand and the diagnostic evaluation should become more comprehensive.
Table 3.1 Familial disorders that can present with young onset dementia
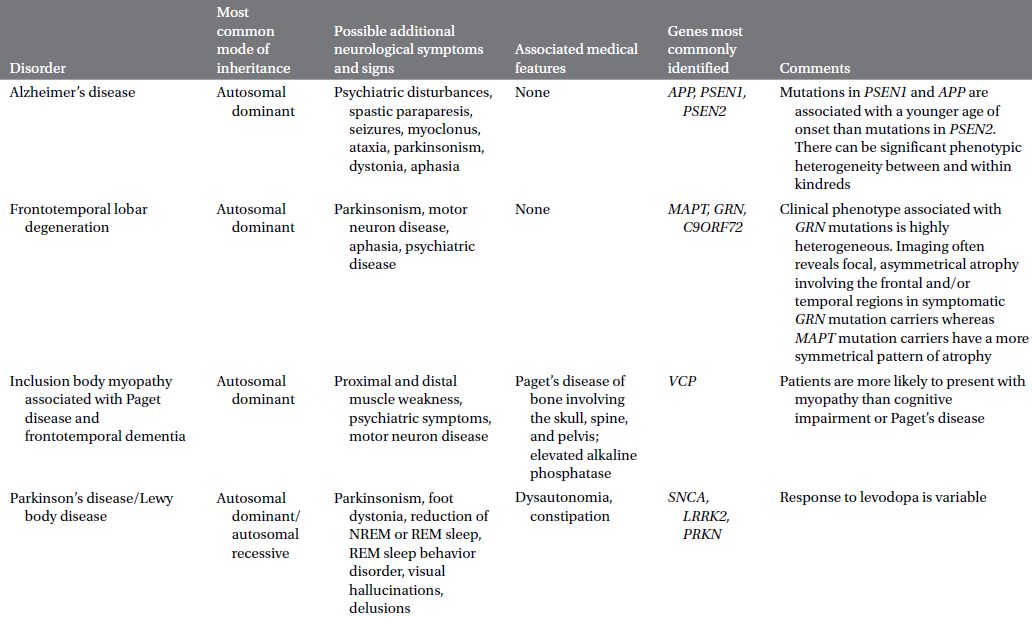

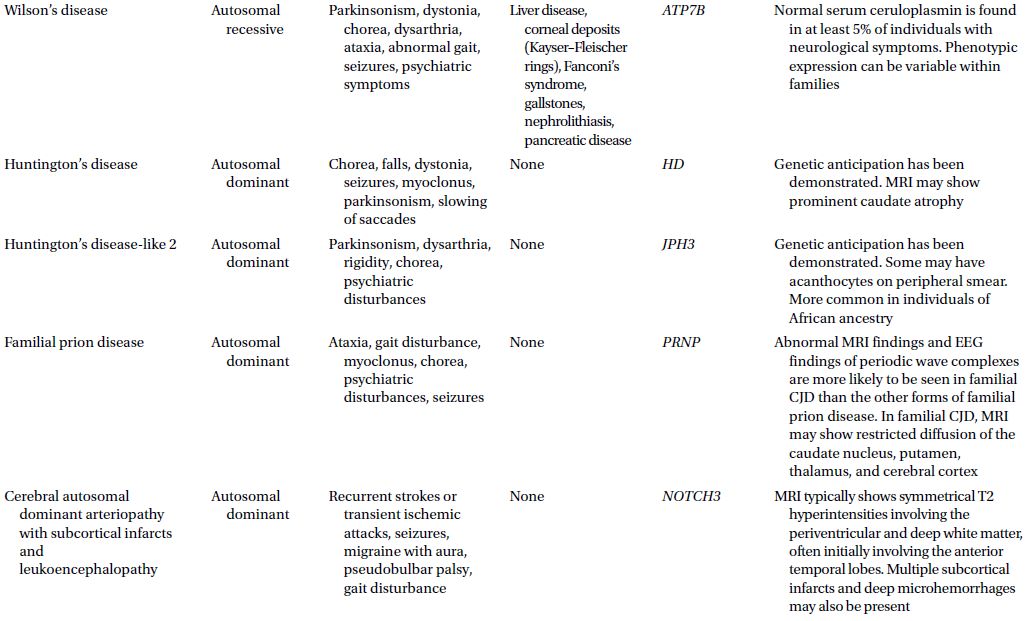
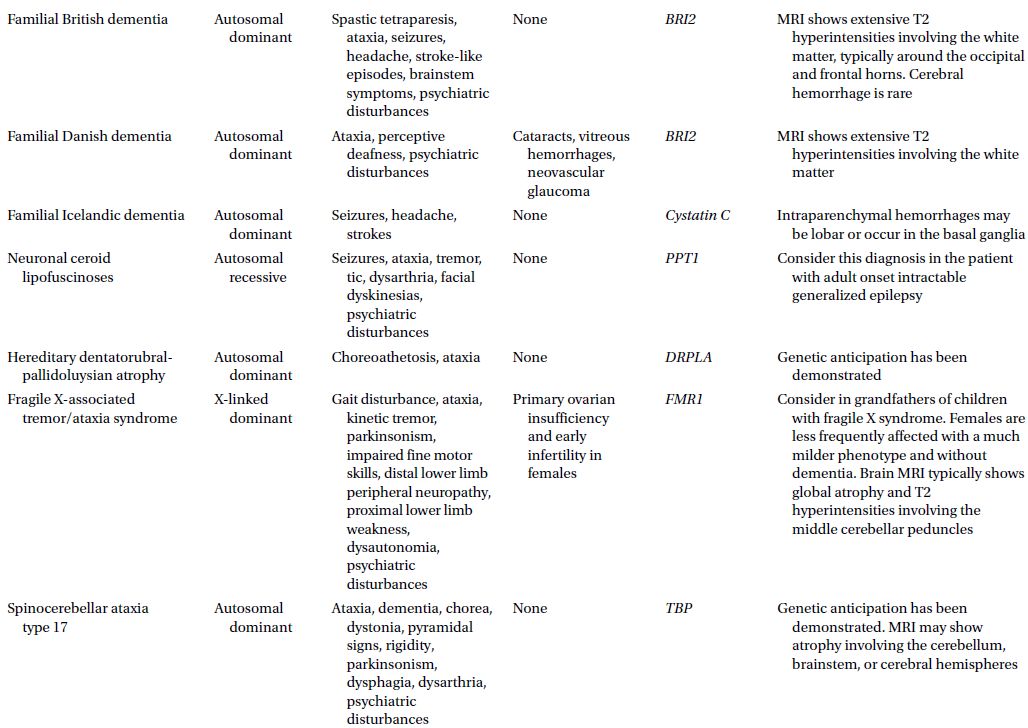
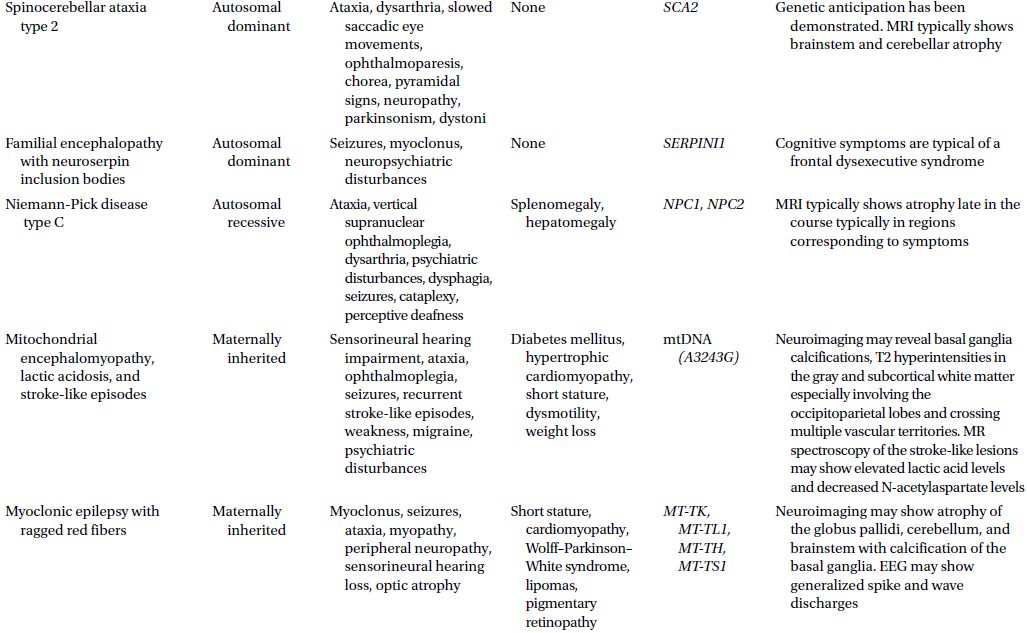
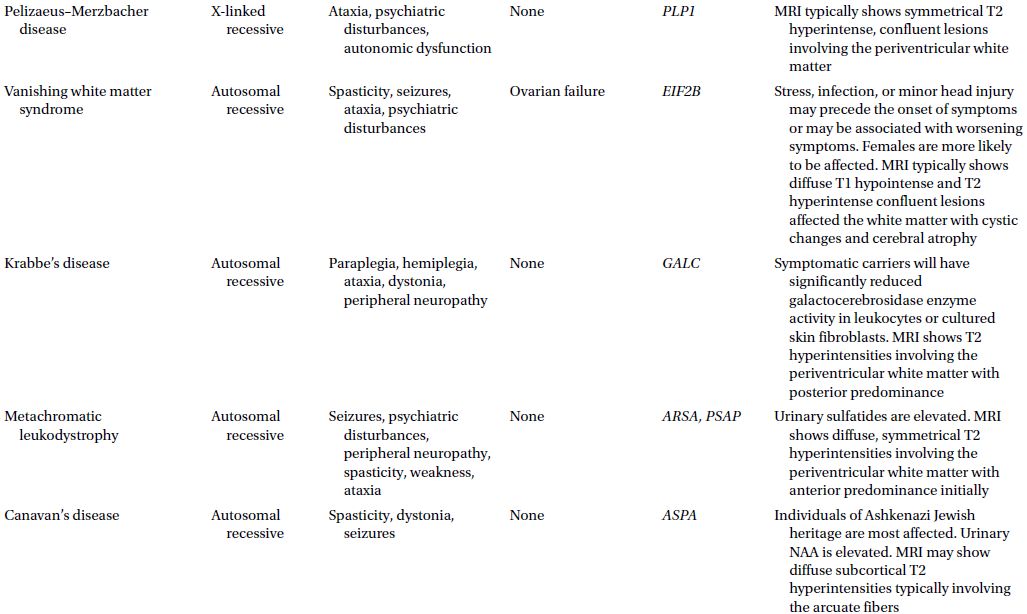


Table 3.2 Etiologies of dementia in cohorts of patients less than 65 years
The initial laboratory testing should serve as a screen for infection or inflammatory conditions, metabolic abnormalities, common nutritional deficiencies, and endocrinopathies.
The complete blood count with leukocyte differential count can rapidly screen for the presence of an inflammatory state, infection, anemia, or malignancy. The presence of normocytic anemia may suggest the existence of gastrointestinal blood loss, poor nutrition, hemolysis, renal insufficiency, primary bone marrow disorder, or anemia of chronic disease, the latter including autoimmune disease, acute or chronic infection, and malignancy. Macrocytic anemia is often associated with alcohol abuse, vitamin B12 deficiency or folate deficiency, if not due to a medication effect. Marked macrocytic anemia (i.e. mean corpuscular volume >110 fL) is due to primary bone marrow disease until proven otherwise. Microcytic anemia can be evidence of iron deficiency, thalassemia, or anemia of chronic disease. Acquired neutropenia may be due to underlying infection or sepsis, drug effect, hematological malignancy, or autoimmune disease (e.g. systemic lupus erythematosus). Acquired lymphopenia can be seen with infection including that from human immunodeficiency virus (HIV), autoimmune disease, connective tissue disease, renal insufficiency, and alcohol abuse. Thrombocytosis may be due to non-myeloid or myeloid malignancy, hemolysis, infection, or chronic inflammatory state. Thrombocytopenia can be seen with thrombotic thrombocytopenic purpura, drug effect, hypersplenism, connective tissue disease, lymphoproliferative disorders, HIV infection, or idiopathic thrombocytopenic purpura.
Sedimentation rate and C-reactive protein are inflammatory markers which can be used as a non-specific screen for malignancy, infection, autoimmune disease, or kidney disease.
Basic metabolic profile which includes electrolytes, creatinine, and blood urea nitrogen should be performed to screen for chronic renal insufficiency and electrolyte derangements. The presence of renal disease and cognitive impairment may suggest a multisystem disorder such as mitochondrial disease, systemic vasculitis (e.g. due to Wegener’s granulomatosis or systemic lupus erythematosus), or sarcoidosis. Renal dysfunction due to diabetes or hypertension can be associated with cerebrovascular disease. Hyponatremia can be a sign of cerebral salt wasting syndrome or syndrome of inappropriate antidiuretic hormone secretion as seen in a number of neurological disorders such as meningitis or encephalitis, including autoimmune or paraneoplastic limbic encephalitis related to voltage-gated potassium antibodies. Many medical conditions can also cause hyponatremia such as congestive heart failure, hepatic failure, renal disease, hypothyroidism, adrenal insufficiency, polydipsia in patients with psychiatric disease, and medication related. Hyponatremic encephalopathy is the most serious clinical manifestation of hyponatremia. The clinical manifestations may include headache, confusion, psychosis, abnormalities of gait, depressed level of consciousness, or seizures. Rapid correction of hyponatremia can result in an osmotic demyelination syndrome in which encephalopathy, parkinsonism, mutism, pseudobulbar palsy, seizures, and locked-in syndrome have all been described.
Screening for an associated endocrinopathy with thyroid function tests, parathyroid hormone, and calcium should be performed early in the diagnostic evaluation. Chronic hypocalcemia, hypothyroidism, hyperthyroidism, hypoparathyroidism, hyperparathyroidism, and hypercortisolism have all been reported to result in psychosis or dementia.
Cognitive and behavioral symptoms can be associated with vitamin B12 deficiency, even in the absence of anemia or macrocytosis, and are often accompanied by sensory loss, paresthesias, and ataxia. A frontotemporal dementia (FTD) syndrome has been described with accompanying frontoparietal and anterior temporal hypoperfusion on brain single photon emission computed tomography (SPECT), with clinical resolution of symptoms and improvement of cerebral perfusion after cyanocobalamin supplementation. If vitamin B12 deficiency is confirmed, then screening for pernicious anemia should occur, with either intrinsic factor antibodies or the Schilling test, before determining that the cause of deficiency is due to primary intestinal malabsorption.
Elevated transaminases and hyperammonemia can be associated with a hepatic encephalopathy which is characterized by altered mentation (e.g. decreased attention, amnesia, confusion) and personality changes (e.g. apathy, depression, euphoria, paranoia) or coma if severe.
Structural neuroimaging, ideally with an magnetic resonance imaging (MRI) study if not contraindicated, should be performed early in the evaluation of YOD. In addition to providing evidence of inflammatory, ischemic, or structural pathology, this can provide information regarding whether a particular pattern of atrophy is present consistent with a specific neurodegenerative disorder. Individuals with Alzheimer’s disease (AD) dementia tend to have diffuse cortical atrophy with more pronounced atrophy involving the hippocampi and posterior temporoparietal lobes, whereas patients with FTD typically have symmetrical or asymmetrical volume loss predominantly affecting the frontal and anterior temporal lobes. On the other hand, dementia with Lewy bodies (DLB) is not typically associated with cortical atrophy but rather atrophy of the dorsal midbrain, substantia innominata, and hypothalamus which may be difficult to visualize on standard MRI sequences. MRI findings in progressive supranuclear palsy are best visualized in the midsagittal plane. Atrophy of the midbrain tegmentum gives a characteristic appearance of a hummingbird in progressive supranuclear palsy. Volume loss of the middle potion of the anterior corpus callosum, anterior cingulate, and frontal cortex, to a lesser degree, may also be seen. Corticobasal syndrome, whether due to corticobasal ganglionic degeneration or another pathological substrate, is typically associated with atrophy of the middle corpus callosum, superior parietal, and posterior frontal lobes.
Coronal MR images should be included as they provide an alternative view for visual inspection of medial temporal lobe structures to assess for atrophy which can be seen in AD and FTD, and also to evaluate for hippocampal sclerosis. Coronal fluid-attenuated inversion recovery (FLAIR) images are crucial in cases of suspected limbic encephalitis as this allows for optimal visualization of hyperintensities involving the medial and basal temporal lobes.
Diffusion-weighted imaging sequences ought to be performed to evaluate for the presence of acute or subacute ischemic infarcts which may be a result of arterial or venous occlusion, primary CNS vasculitis, or secondary vasculitides from infection, collagen vascular disease, or drugs. Restricted diffusion may also be seen with pyogenic abscess, acute demyelination, tumor with a high cellularity, or osmotic demyelination syndrome. Hypoglycemia, hypoxic-ischemic insult, and carbon monoxide poisoning each result in a characteristic pattern of restricted diffusion as well. Individuals with Creutzfeldt–Jakob disease (CJD) may have restricted diffusion involving the striatum, thalamus, and/or cortical ribbon.
The presence of multiple microhemorrhages detected on gradient echo MR sequences should alert the physician to possible cerebral amyloid angiopathy, which can be associated with AD, or chronic hypertension. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), diffuse axonal injury associated with traumatic head injury, and CNS vasculitis are less common causes of cerebral microhemorrhages. Gradient echo sequences can more easily identify brain iron deposition as can be seen in neuroferritinopathy, pantothenate kinase-associated neurodegeneration, and aceruloplasminemia. Bilateral hypointensities of the globus pallidi on gradient echo sequences are also associated with chronic hepatic encephalopathy which is felt to most likely reflect deposition of manganese and correspond to hyperintensities on T1 sequences.
White matter disease is typically best seen on FLAIR images and can present with a variety of cognitive symptoms. The differential in the young patient with dementia and an MRI consistent with a leukoencephalopathy can be quite broad and includes dysimmune, infectious, postinfectious, vascular, metabolic, nutritional, neoplastic, toxic, demyelinating, or mitochondrial disorders. Administration of contrast may help to narrow the possibilities. The adult onset leukodystrophies have a more heterogeneous presentation than the childhood forms and can present acutely or with a slowly progressive course spanning decades. Disorders of the urea cycle, fatty acid oxidation, or amino acid catabolism may present with a progressive dementing syndrome accompanied by spastic paraparesis and psychiatric disturbances often exacerbated by stressors such as minor trauma, pregnancy, or infection. These individuals may be misdiagnosed as having multiple sclerosis or a primary psychiatric disorder.
If the imaging study reveals evidence of “brain sagging” with swelling of brainstem structures and low-lying cerebellar tonsils, then additional studies such as CT myelography or heavily T2-weighted MR myelography should be pursued to rule out a CSF leak. Brain sagging has been described to cause a behavioral variant FTD phenocopy which has been referred to as frontotemporal brain sagging syndrome. Pachymeningeal enhancement should prompt further evaluation with a lumbar puncture to measure the opening pressure as low pressure can give this picture. In addition, CSF analysis to evaluate for evidence of inflammation, infection, and malignancy may be necessary.
Round 2
If the above laboratory and imaging studies are non-diagnostic and the patient and family are interested in pursuing further testing to arrive at a diagnosis, then the next round of testing should follow.
Autoimmune and collagen vascular diseases should be considered in all patients with YOD, but especially in those with onset of dementia shortly following pregnancy, family history or personal history of preexisting autoimmune disease, signs or symptoms suggestive of underlying collagen vascular disease (e.g. uveitis, arthritis, sicca syndrome, unexplained fevers, or recurrent oral or genital ulcers). Screening labs can include serum antinuclear antibody, rheumatoid factor, anti-Ro and anti-La antibodies, antiphospholipid antibodies (i.e. lupus anticoagulant, anticardiolipin antibodies, and anti-beta2-glycoprotein-1 antibodies), angiotensin converting enzyme, and autoantibodies to thyroid peroxidase and thyroglobulin. A pathergy test should be performed if Behçet’s disease is being considered.
Serological testing for Treponema pallidum, HIV, and Borrelia burgdorferi should be performed early to screen for these infectious etiologies for which there are treatments.
An elevation in serum amylase can be associated with pancreatic encephalopathy which can manifest as cognitive impairment, focal neurological signs, and psychosis.
Neuropsychological assessment is a tool that can be used to demonstrate subtle cognitive impairment that may be difficult to detect on the bedside mental status exam. In addition, it can provide valuable information on the extent and severity of cognitive dysfunction. A particular pattern of cognitive impairment may become evident and assist in making the diagnosis of a specific neurodegenerative disorder.
An EEG is a non-invasive tool that in this setting may be pursued if non-convulsive status epilepticus or subclinical seizures are suspected, or in the patient with a rapidly progressive dementia. Bilateral, generalized, or unilateral periodic bi- or triphasic sharp wave complexes is a pattern suggestive of CJD in the appropriate clinical context and is typically seen in certain subtypes. Serial EEG recordings may be required if suspicion for CJD is high. Seizures, including those refractory to anticonvulsants, have been associated with paraneoplastic and non-paraneoplastic limbic encephalitis as well as a steroid-responsive encephalopathy associated with autoimmune thyroiditis. Encephalopathy is usually accompanied by slowing of the posterior dominant rhythm to a degree correlated with the extent of cortical dysfunction, but is an otherwise non-specific finding. Subacute sclerosing panencephalitis (SSPE), which is more common in underdeveloped and developing countries due to the lower rates of measles immunization, is rarely seen in adults. However, there is growing concern that its incidence will increase in developed countries in the face of possible decline in vaccination due to unconfirmed claims regarding increased risk of autism. EEG in SSPE typically shows periodic, bisynchronous, high-amplitude slow waves occurring at long repetition intervals.
An electrocardiogram can be performed as a screen for cardiomyopathy or conduction defects seen in HIV, Lyme disease, mitochondrial, lipid, and amino acid disorders. Chest x-ray can be performed to screen for lung cancer, pulmonary tuberculosis, or sarcoidosis.
Cerebrospinal fluid examination should include cell count with differential, protein, and glucose. Measurements of IgG index and oligoclonal bands should be performed in all cases to evaluate for the presence of an immunological response. Evaluation for chronic meningoencephalitis should include polymerase chain reaction (PCR) for Tropheryma whipplei, Lyme serology, India ink stain, culture for Cryptococcus neoformans, acid-fast bacilli smear and culture for tuberculosis, and PCR for herpes simplex virus type 2. An elevated level of neuron-specific enolase, 14-3-3, and/or tau is indicative of rapid neuronal death and can be used as adjunctive tests in the diagnosis of CJD. A low CSF amyloid beta 1–42 and high total tau or phosphorylated tau is suggestive of AD pathology with good sensitivity and specificity. In the immunocompromised patient, CSF analysis should include PCRs for human herpesvirus-6, human herpesvirus-7, JC virus, and cytomegalovirus. Further microbial studies should be guided by the results of neuroimaging and initial CSF studies.
Evaluating for the presence of a paraneoplastic antibody is reasonable in all patients in whom the initial work-up is unrevealing. If serum paraneoplastic antibodies are negative and clinical suspicion is high, then the CSF should be tested as there are cases in which an autoantibody may only be detected in the CSF. Testing for an occult malignancy should parallel the search for a paraneoplastic antibody. Whole-body positron emission tomography using [18 F] fluorodeoxyglucose (FDG-PET) has a significantly higher sensitivity than CT alone in detecting occult malignancy. A PET scan may also identify granulomatous disease. Given the cost of a whole-body PET scan, it is reasonable to begin with CTs of the chest, abdomen, and pelvis, mammography in women, prostate-specific antigen in men, and/or testicular ultrasound in young men. A transvaginal ultrasound is the preferred imaging modality for detecting primary adnexal malignancy in females, followed by an MRI with contrast if a mass is identified. The imaging studies performed may also be guided by the specific antibody detected.
Round 2.5
Alternative diagnostic studies may be necessary based on the patient’s clinical presentation, co-morbidities, exam findings, or results of testing performed early in the evaluation.
For patients in whom malnutrition is suspected (e.g. those with end-stage liver or renal disease, chronic obstructive lung disease, malabsorption syndrome, alcoholism, malignancy, cystic fibrosis, short bowel syndrome, or prior bariatric surgery), in addition to electrolyte and vitamin B12 levels, screening for other nutritional deficiencies should be considered. Niacin (vitamin B3) deficiency causes a constellation of symptoms known as pellagra in which a painful dermatitis involving skin exposed to sun or pressure, diarrhea, psychiatric disturbance, and dementia are present. Measurement of erythrocyte transketolase activity can serve as an indicator of thiamine status, deficiency of which is associated with Wernicke–Korsakoff syndrome. Serum vitamin E level should be checked as severe deficiency, whether due to acquired or familial etiologies, can present with progressive neurological deterioration marked by peripheral neuropathy, ataxia, myopathy, and dementia. Copper, retinol (vitamin A), zinc, biotin, and vitamin B6 are each rarely associated with a potentially reversible encephalopathy and serum levels should be measured in the appropriate clinical circumstance.
If neuroimaging shows evidence of previous infarcts, the underlying etiology should be identified, beginning with evaluating for typical cerebrovascular risk factors with lipid profile, fasting glucose or glycosylated hemoglobin, hypercoagulable panel, and echocardiogram. In addition to the connective tissue screening laboratory studies mentioned above, serum antineutrophil cytoplasmic antibodies and cryoglobulins may be diagnostic. An underlying mitochondrial disorder may also need to be pursued.
In the patient with an accompanying movement disorder such as chorea, parkinsonism, or dystonia, serum ferritin, copper, ceruloplasmin, and 24-h urinary copper should be measured. The peripheral blood smear can provide additional information. Acanthocytes are associated with a number of neurodegenerative disorders including chorea-acanthocytosis, Huntington’s disease-like 2, and pantothenate kinase-associated neurodegeneration. A slit-lamp examination should be performed to evaluate for the presence of Kayser–Fleischer rings. The presence of ataxia should prompt measurements of serum vitamin E, vitamin B12, copper, IgA endomysial and tissue transglutaminase antibodies, and paraneoplastic antibodies.
An overnight polysomnogram can be performed if obstructive sleep apnea or rapid eye movement (REM) sleep behavior disorder is suspected. REM sleep behavior disorder has been associated with limbic encephalitis and preceding or accompanying DLB. Insomnia and narcolepsy are each also associated with limbic encephalitis.
If clinical suspicion is present, electromyogram and nerve conduction studies should be performed to evaluate for co-existing motor neuron disease, myopathy, or peripheral neuropathy.
Round 3
Functional imaging of the brain using FDG-PET is a reliable and valid diagnostic tool that can provide additional data early in the investigation to support the diagnosis of a specific neurodegenerative disorder, but should not be interpreted in isolation of the clinical picture. A pattern of hypometabolism involving the posterior temporal and posterior parietal lobes, including the posterior cingulate and precuneal regions, with sparing of the primary visual, sensory, and motor cortices, is classic for AD dementia. In FTD, hypometabolism is more often seen in anterior temporal, anterior cingulate, and frontal regions. Patients with DLB tend to have similar patterns of metabolic reductions as those of patients with AD, with significant hypometabolism involving the posterior cingulate and parietotemporal regions, but with additional hypometabolism involving the primary visual cortex. Caudate and putamen hypometabolism are seen in Huntington’s disease, even in early stages.
If a mitochondrial cytopathy is suspected, then serum and CSF lactate and pyruvate should be measured, but negative results do not rule out the diagnosis. A fasting or postexercise sample of CSF and serum lactate may increase the sensitivity. Quantification of urine and plasma amino acids should identify an amino acid disorder and an elevated alanine may be suggestive of a mitochondrial cytopathy. Measurement of urine organic acids and blood spot acyl carnitines can identify an organic acid or mitochondrial disorder. Serum ammonia level should be checked to screen for a urea cycle disorder in the encephalopathic patient. An elevated creatine kinase may be indicative of a mitochondrial disorder as well.
Enzyme assays on leukocytes or cultured skin fibroblasts can also be performed to diagnose some of the lysosomal storage disorders. Accumulation of the stored material due to the enzyme defect may be measured in the tissue or urine (e.g. N-substituted amino acid in Canavan’s disease and sulfatides in metachromatic leukodystrophy).
In addition to screening for possible mitochondrial or lipid storage disease in the young patient with episodic cognitive or behavioral symptoms, screening for porphyria should be considered and includes measurements of urinary porphobilinogen, ideally when the patient is symptomatic. If the screening test is negative but there is strong suspicion, further testing should proceed with quantification of porphobilinogen, delta-aminolevulinic acid, and total porphyrins from a 24-h urinary collection.
If adrenomyeloneuropathy is suspected, serum very long chain fatty acids should be measured.
Mercury, arsenic, lead, manganese, tin, thallium, and aluminum are some of the heavy metals for which chronic exposure can result in dementia. A 24-h urinary collection can be used to confirm toxicity for arsenic, mercury, thallium, or tin. Consumption of seafood should be avoided for 3 days prior to testing as it can raise urinary arsenic levels. Lead and manganese should be measured in the blood to demonstrate toxicity. Elevation in the blood level of aluminum following deferoxamine infusion is diagnostic of aluminum toxicity. Basophilic stippling of red blood cells is seen on peripheral blood smear in cases of lead, aluminum, or arsenic toxicity.
Referral to colleagues in other medical departments may be necessary to find evidence of systemic involvement of a disorder. An ophthalmological examination may provide valuable information. Optic atrophy and/or retinitis pigmentosa are associated with mitochondrial or lysosomal storage disease. Retinal degeneration, corneal clouding, and a cherry-red spot are each associated with storage diseases. Cataracts can be associated with some leukodystrophies or cerebrotendinous xanthomatosis. Ocular and optic nerve changes are seen in paraneoplastic disorders and immune-mediated disease such as Behçet’s disease, sarcoidosis, and systemic lupus erythematosus. A slit-limp examination should be included to identify Kayser–Fleischer rings associated with Wilson’s disease. In patients in whom metabolic or mitochondrial disorders are being considered, colleagues in pediatric neurology can be instrumental.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


