
CN, cranial nerve; CST, corticospinal tract; ICP, inferior cerebellar peduncle; LST, lateral spinothalamic tract; ML, medial lemniscus; PICA, posterior inferior cerebellar artery; SCM, sternocleidomastoid; ST, solitary tract.
Wallenberg described the LMS, the most common form of brainstem stroke. Wallenberg’s original patient had an occlusion of the PICA, but LMS is most often due to ischemia in the PICA distribution because of vertebral artery occlusion (Figure 21.1). Typical manifestations include vertigo, nausea and vomiting, nystagmus, hoarseness, dysphagia, dysphonia, singultus, ipsilateral hemiataxia, and numbness of the ipsilateral face and contralateral body. Occipital headache or pain in the back of the neck may occur at the onset; prominent pain raises the possibility of vertebral artery dissection. The patient may be unable to talk and swallow initially. Clinical findings are summarized in Table 21.1.
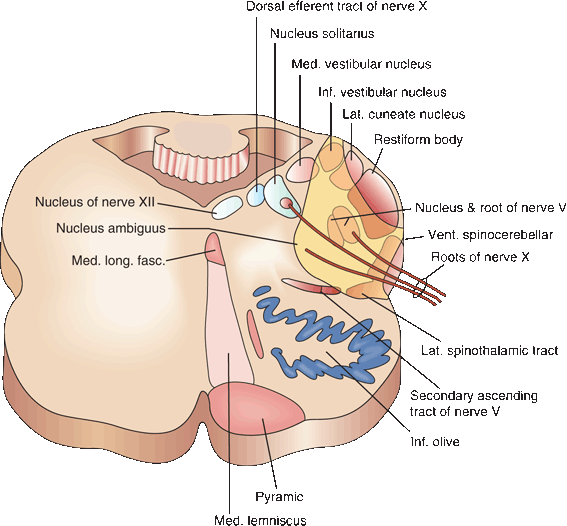
FIGURE 21.1 Cross section of the medulla illustrating the site of the lesion following thrombosis of the posterior inferior cerebellar artery.
In a series of magnetic resonance imaging (MRI)–verified LMS, the most common findings were ipsilateral Horner’s syndrome and ataxia, and contralateral body hypalgesia. The spontaneous nystagmus is usually horizontal or mixed horizontal-torsional. Horizontal nystagmus beats away from the side of the lesion and may be second or third degree. Torsional nystagmus with the upper poles beating away from the side of the lesion is also common. The nystagmus is influenced by head and eye position and by fixation. Dysphagia is common. It is often more severe than would be expected simply from a lesion of the nucleus ambiguus, and disruption of connections to a premotor swallowing center in the dorsolateral medulla has been postulated. Partial resolution and survival is the rule; the ability to swallow and talk returns, although residual hoarseness, persistent ataxia, and sensory changes may remain. Aspiration is a major threat. The presence of dysphonia, soft palate dysfunction, or facial sensory loss suggests an increased risk. Although LMS is usually ischemic, it has also been described with aneurysm, abscess, hematoma, arteriovenous malformation, demyelinating disease, and metastatic neoplasm. The LMS may have many unusual manifestations (Box 21.2).
Unusual Manifestations of Lateral Medullary Syndrome
Patients may have an ipsilateral upper motor neuron facial palsy due to involvement of Dejerine’s aberrant pyramidal tract (see facial nerve chapter). In the series of Sacco et al., mild ipsilateral facial weakness was present in 42% of patients, usually limited to the lower face. The hypalgesia may involve only the ipsilateral face or only the contralateral body; the classical crossed pattern occurs only in a minority. Other patterns of sensory loss are discussed in Chapter 15. Ocular motor abnormalities are common, including skew deviation with ipsilateral hypotropia, ocular tilt reaction, bizarre environmental tilt illusions including world inversion (floor on ceiling phenomenon), ipsilateral gaze deviation with impaired contralateral pursuit, saccadic abnormalities, see-saw nystagmus and eyelid nystagmus. Ocular abnormalities and facial weakness are common and do not imply extension of the lesion beyond the lateral medulla. There may be contralateral hemiparesis due to inferior extension of the zone of ischemia to the medullary pyramid prior to decussation or ipsilateral hemiparesis due to inferior extension to the lateral funiculus of the rostral spinal cord (Opalski’s sub-medullary syndrome, Figure 21.2). Rarely, there is impaired sensation of the ipsilateral arm and leg due to inferior extension to the gracile and cuneate nuclei, ipsilateral loss of taste or contralateral facial hypalgesia. Other unusual manifestations include wild unilateral, proximal arm ataxia; neurotrophic ulceration of the face; inability to sneeze; paroxysmal sneezing; loss of taste; Ondine’s curse; and weakness of the sternocleidomastoid. Chronic central facial pain develops in some patients.

FIGURE 21.2 Imaging features of Opalski syndrome. Fluid-attenuated inversion recovery MRI. A. Directionally encoded map with hues reflecting tensor orientation B. Superimposed images. C,D. A yellow halo represents the infarct and blue lines the pyramidal tracts (coronal); the tracts fuse at the decussation (transverse). Caudal extension of the lesion involves the ipsilateral corticospinal tract (arrows) after the decussation (arrowheads). (Reprinted from Nakamura S, Kitami M, Furukawa Y. Opalski syndrome: ipsilateral hemiplegia due to a lateral-medullary infarction. Neurology 2010;75:1658, with permission.)
ANATOMIC BRAINSTEM SYNDROMES
The other approach to organizing brainstem syndromes is by the anatomical area or the major blood vessel involved. The midbrain syndromes are variations and combinations of an ipsilateral third nerve palsy and weakness, ataxia, or tremor of the contralateral limbs; this is due to ischemia in the distribution of paramedian penetrating vessels from the rostral basilar artery. The LMS is discussed in the previous section, and the medial medullary syndrome in Box 21.1.
The vascular pontine syndromes can be divided into medial and lateral and into superior, middle, and inferior. The medial pontine syndromes are due to disease of the paramedian perforators; the lateral pontine syndromes are due to disease of the circumferential arteries. The AICA supplies the lateral inferior pons and upper medulla, whereas the SCA supplies the lateral upper pons. The midpons is supplied by a short circumferential artery. Just as PICA ischemia causes the LMS, ischemia in the AICA distribution causes the lateral inferior pontine syndrome; and ischemia in the SCA distribution causes the lateral superior pontine syndrome. The generally recognized pontine syndromes are therefore the medial inferior pontine, lateral inferior pontine (AICA), medial and lateral midpontine, medial superior pontine, and lateral superior pontine (SCA). The vascular pontine syndromes are summarized in Table 21.2. In a series of patients with lesions involving the AICA distribution, only 29% had the complete AICA syndrome. Partial syndromes were characteristic of small vessel disease; more widespread involvement indicated basilar artery occlusive disease. The SCA syndrome is also often partial. Basilar branch occlusion may involve any of the branches of the basilar artery. The mechanism is atherothrombotic occlusion at the point of origin of the branch, and the infarction typically extends to the ventral surface of the pons.
TABLE 21.2 Summary of the Brainstem Syndromes Organized by Anatomical Region and Blood Vessel Involved

AICA, anterior inferior cerebellar artery; CN, cranial nerve; CST, corticospinal tract; CTT, central tegmental tract, ICP, inferior cerebellar peduncle; INO internuclear ophthalmoplegia; LST, lateral spinothalamic tract; MCP, middle cerebellar peduncle; ML, medial lemniscus; MLF, medial longitudinal fasciculus; PPRF, pontine paramedian reticular formation; SCA, superior cerebellar artery; SCP, superior cerebellar peduncle; ST, solitary tract.
Vertebrobasilar transient ischemic attacks (vertebrobasilar insufficiency, VBI) are episodes of brainstem ischemia due to occlusive disease involving the posterior circulation. Symptoms depend upon which region of the brainstem is ischemic. The clinical manifestations of an attack of VBI are typically bilateral, with varying degrees of weakness, numbness, and CN dysfunction. Accompanying symptoms indicative of brainstem dysfunction include diplopia, dysarthria, dysphagia, vertigo, nausea, and vomiting. There may be impaired vision due to ischemia in the posterior cerebral artery distribution. Bilateral sensory complaints are common, especially circumoral paresthesias. Attacks usually last from a few minutes to half an hour, sometimes longer.
Basilar artery occlusion may have a gradual onset or a fluctuating course with prodromata, but often the symptoms appear apocalyptically; death may occur within a short period of time. When the onset is acute, there is sudden loss of consciousness with gradually increasing coma and flaccid extremities or decerebrate rigidity. The onset may be subacute with prodromal vertigo, nausea, headache, and paresthesias, which may occur up to 2 weeks before the stroke, followed by bilateral CN and long tract abnormalities (progressive basilar thrombosis). Fisher described a “herald hemiparesis” in basilar artery thrombosis, frequently present at an early stage, when brainstem signs are absent or inconspicuous, followed within a few hours by bilateral hemiplegia and coma or a locked-in state (Chapter 51). With total occlusion, there is either hemiplegia on one side and partial hemiplegia on the other, or quadriplegia. Pseudobulbar palsy with severe dysphagia and dysarthria result from bilateral involvement of the supranuclear fibers to the medullary nuclei. Involvement of ascending sensory pathways causes a disturbance of both deep and superficial sensations on the body, extremities, and sometimes the face. The pupils are usually miotic and poorly reactive. Ocular bobbing and palatal myoclonus may occur. The neurologic signs are characteristically variable and complex. Coma and decerebrate rigidity with respiratory and circulatory instability is common. Patients with coma at the outset have a grave prognosis. The pathogenesis in younger patients is usually cardiac embolism or vertebral artery dissection; in elderly patients, local atherothrombosis is more common. Many patients have a history of hypertension, diabetes mellitus, and cerebrovascular disease. The site of occlusion is usually in the lower third of the basilar artery. The outcome with severe brainstem ischemic disease is usually poor. Death is a common outcome of complete basilar artery occlusion. Patients may be left in a locked-in state (Chapter 51).
The “top of the basilar” syndrome is caused by ischemia in the distribution of the distal basilar artery, usually embolic, involving the rostral brainstem, thalamus, and portions of the cerebral hemispheres fed by the posterior cerebral arteries. A variety of oculomotor and pupillary abnormalities may occur, along with visual and behavioral abnormalities, often without significant extremity weakness.
Hemorrhage into the brainstem, especially the pons, is common, particularly in patients with hypertension. Patients with pontine hemorrhage have a clinical picture similar to basilar artery occlusion, but warning symptoms are less apt to occur. They are comatose, quadriplegic, have bilateral facial paralysis, bilateral horizontal gaze palsies, and pinpoint poorly reactive pupils. Hyperthermia is common. Imaging studies often show a large hematoma in the midpons. Few patients survive such an event. The initial level of consciousness and the size of the hematoma are strongly related to the outcome. Coma within 2 hours of onset and a transverse diameter of the hematoma on computed tomography of more than 2 cm indicate a poor prognosis. Smaller hematomas produce less dramatic deficits, and hemipontine syndromes may occur.
Pressure on the brainstem due to supratentorial mass effect can cause either lateral transtentorial herniation (uncal syndrome), with third nerve involvement and signs of lateral midbrain compression, or central transtentorial herniation, with constricted pupils, Cheyne-Stokes respirations, bilateral corticospinal tract signs, decorticate rigidity, and progressive impairment of diencephalic, midbrain, pontine, and medullary function. Because of the patterns of venous drainage, increased intracranial pressure and herniation at either the foramen magnum or the tentorium may cause secondary bleeding into the midbrain, pons, or medulla. Duret hemorrhages are secondary hemorrhages into the upper brainstem that occur with increased intracranial pressure and descending transtentorial herniation. Brainstem hemorrhage may cause hyperthermia, respiratory abnormalities, coma, and finally death in patients with brain tumors, subarachnoid hemorrhage, cerebral hemorrhage, trauma, rapidly expanding supratentorial mass lesions, or similar conditions causing an increase in intracranial pressure. Affected patients rarely survive; Stiver et al. reported an exception in a young adult traumatic brain injury patient.
When increased intracranial pressure causes tonsillar herniation, the cerebellar tonsils and lower medulla are forced downward through the foramen magnum. Although tonsillar herniation is a feared complication of lumbar puncture done in the face of increased intracranial pressure, it is in fact rare. Medullary compression causes profound impairment of all vital functions, with bradycardia, either a fall or rise in blood pressure, slow or rapid respirations, soaring temperature, convulsions, unconsciousness, and death. The Cushing (vasopressor) reflex (response, reaction, or effect) is hypertension, increased pulse pressure, bradycardia, and slow, irregular respirations seen in patients with increased intracranial pressure and brainstem compression. The full triad occurs in only about one-third of cases, and some patients may have isolated hypertension. On postmortem examination a pressure cone may be seen on the medulla.
Aneurysms of the basilar or vertebral arteries or their branches, and hemangiomas, may cause extramedullary compression and CN involvement. Arteriovenous malformations may cause intramedullary or extramedullary dysfunction, depending on their extent and location. Extravasation of blood about the base of the brain from subarachnoid or intracerebral hemorrhage may affect the CNs as they leave the skull.
Lacunes are small, deep infarctions in the territory of a deep penetrating arteriole. Hypertension is the major predisposing factor. The brainstem, particularly the pons, is a common location for lacunar infarction. Lacunar syndromes due to brainstem involvement include pure motor stroke, dysarthriaclumsy hand syndrome, and ataxic hemiparesis (homolateral ataxia and crural paresis). In a study of 37 patients with acute infarcts mainly involving the base of the pons, pure motor hemiparesis was present in 17, sensorimotor stroke in 3, ataxic hemiparesis in 4, and dysarthria-clumsy hand syndrome in 6 patients. The pathogenetic mechanisms of ischemia were lacunar events or basilar atheromatous branch occlusion in most. Large lesions involving the para-median caudal or middle pons were more likely to cause pure motor stroke, and lesions in the paramedian rostral pons tended to produce dysarthria-clumsy hand syndrome. The different pontine lacunar syndromes reflect the balance of involvement of the corticospinal, corticopontocerebellar, and corticobulbar tracts.
Other unusual, typically vascular, brainstem syndromes are briefly described in Box 21.3.
Other Brainstem Syndromes
The one-and-a-half syndrome is a horizontal gaze palsy and ipsilateral internuclear ophthalmoplegia, or INO (Chapter 14). The association of an ipsilateral lower motor neuron facial nerve palsy and a one-and-a-half syndrome has been termed the eight-and-a-half syndrome. The Brissaud-Sicard syndrome is ipsilateral hemifacial spasm and contralateral hemiparesis due to a pontine lesion. The lateral pontomedullary syndrome consists of the findings of the lateral medullary syndrome with additional involvement of CNs VII and VIII consistent with extension of the lesion to the inferior pons. Raymond-Cestan syndrome is horizontal or vertical gaze palsy, contralateral hemiparesis or quadriparesis, hemianesthesia, and athetosis due to basilar branch occlusion. Rasdolsky’s syndrome is contracture and paresis of the masseter and facial muscles due to neoplasm of ipsilateral pontine tegmentum. Marie-Foix syndrome is contralateral hemiparesis and hypalgesia with ipsilateral cerebellar ataxia due to a lesion involving the lateral pons. Other unusual manifestation of brainstem disease include pontine anosognosia, cognitive dysfunction, painful isolated Horner’s syndrome, head shaking nystagmus, jaw opening dystonia, hemidystonia, facial pain syndromes, a sensory level on the trunk, unilateral hyper-or hypohidrosis, upside-down reversal of vision, tonic seizures, and convulsive-like movements.
Nonvascular Brainstem Disorders
Brainstem gliomas are astrocytomas that diffusely infiltrate the brainstem. They occur primarily in children, but adults are occasionally affected. The degree of malignancy varies from low grade to highly anaplastic. Most involve the pons, but any level of the brainstem may be affected. There is typically a combination of multiple cranial nerve palsies (MCNPs), gaze palsy, long tract signs, and ataxia. Because of the slow evolution, there is sometimes a paucity of neurologic signs in spite of the size of the tumor. The lesions may be exophytic, with tumor outgrowth extending beyond the normal limits of the brainstem. If ventricular obstruction occurs, there may be hydrocephalus and increased intracranial pressure. Ependymomas and medulloblastomas may also involve the brainstem. Extramedullary tumors (neurofibromas, schwannomas, meningiomas, hemangiomas, metastases) may cause pressure effects. The course of a brainstem neoplasm is progressive. Increased intracranial pressure may appear late, particularly in brainstem gliomas. Extrinsic metastases and neoplasms that spread by direct extension from the nasopharynx and neighboring sites may cause widespread CN involvement and bone erosion with signs of brainstem compression. Tuberculomas, sarcoidosis, and other granulomas may produce a picture similar to neoplasms.
Brainstem encephalitis (Bickerstaff’s encephalitis) is a clinical syndrome of acute diffuse or multifocal brainstem dysfunction with cerebrospinal fluid (CSF) pleocytosis and increased protein. Actual viral infection has seldom, if ever, been documented, and the disease is usually immunologically mediated. Patients develop ophthalmoplegia and ataxia followed by gradual brainstem dysfunction and altered consciousness. The illness is usually preceded by a viral infection. Some patients have serum anti-GQ1b IgG autoantibodies, the same antibody found in Miller Fisher syndrome (ophthalmoplegia, ataxia, and areflexia). Bickerstaff’s brainstem encephalitis is not to be confused with Bickerstaff’s (basilar artery) migraine (see below). Brainstem encephalitis may be paraneoplastic. Rhombencephalitis refers to inflammatory disease affecting the hindbrain (brainstem and cerebellum). It has a wide variety of etiologies, including multiple sclerosis (MS), Behcet’s disease, paraneoplastic syndrome, lupus and viral and tuberculous infection. Listeria monocytogenes is particularly likely to cause rhombencephalitis; it accounted for 9% of cases in one series.
Demyelinating disease frequently involves the brainstem. INO due to a demyelinating lesion involving the MLF is a very common clinical manifestation of MS. MS can cause lesions elsewhere in the brainstem and can occasionally simulate one of the vascular syndromes. Acute disseminated encephalomyelitis may affect the brainstem, and the involvement is occasionally limited to the brainstem.
In central pontine myelinolysis (osmotic demyelination syndrome), there is widespread, symmetric myelin loss in the central portion of the pons. Lesions commonly occur in other sites as well (extrapontine myelinolysis). Central pontine myelinolysis occurs especially in alcoholics or other malnourished or debilitated individuals and after correction of severe hyponatremia. It typically begins with diplopia, dysphagia, dysarthria, and other evidence of brainstem dysfunction, followed by quadriplegia, mutism, and extensor rigidity. Central pontine myelinolysis runs a fulminating course and is often fatal.
Developmental or congenital anomalies of the craniocervical junction are frequently associated with brainstem dysfunction. The bony walls of the foramen magnum and upper spinal canal lie in close anatomic relationship to the lower brainstem, upper spinal cord, and cerebellum. Neurologic abnormalities may be produced by mechanical compression by the bony abnormality, but often the bony abnormality and the neural abnormality are part of the same process. Platybasia, basilar impression, occipitalization of the atlas, and cervical spina bifida are examples of primary bony abnormalities. Klippel-Feil syndrome is the congenital fusion of two or more cervical vertebrae. There may be accompanying craniocervical junction abnormalities. The associated neurologic abnormalities may include myelopathy, radiculopathy, syringomyelia, and mirror movements.
Arnold-Chiari (or simply Chiari, who made the greater contribution) malformation is a congenital maldevelopment of the brainstem and cerebellum. The cerebellar tonsils are herniated or displaced down into the upper cervical spinal canal. With more severe maldevelopment, the inferior vermis, lower medulla, and fourth ventricle may also be displaced below the foramen magnum. Clinical manifestations include headache, cerebellar ataxia, nystagmus (typically downbeat), and other brainstem deficits. Three varieties commonly occur. Type 1 is the hindbrain malformation only; it can present in adulthood. Mild type 1 Chiari malformations are not uncommonly found on MRI imaging done for other reasons and may be totally asymptomatic. Type 2 is a more severe hindbrain defect usually associated with a lumbar meningomyelocele. Type 3 is the same as type 2 except that the meningomyelocele or encephalocele occurs in the occipitocervical region. The Dandy-Walker syndrome is agenesis of the cerebellar vermis with a massively dilated fourth ventricle forming a cystic structure that occupies most of the posterior fossa. A fourth type of Chiari malformation (cerebellar hypoplasia) is sometimes included; it is the same as the Dandy-Walker cyst.
Syringobulbia is a slit-like cavity in the brainstem. A brainstem syrinx is usually a rostral extension of a syringomyelic cavity from the cervical spinal cord in a patient with a type 1 Chiari malformation, but syringobulbia may rarely occur de novo. MRI detection of syringomyelia has led to earlier treatment and prevention of upward extension of the cavity, and the condition is encountered less frequently now than in the past. In syringobulbia, the syrinx most often involves the lateral medullary tegmentum. The cavity is usually restricted to the lower brainstem but may extend to the pons and rarely higher. The cavity and the resultant clinical picture are typically asymmetric, with lower CN dysfunction, facial numbness, and nystagmus. The facial sensory loss may be in an onion-skin distribution, initially sparing the nasal tip and perioral region. Hypoglossal weakness and atrophy may occur. Facial myokymia is an unusual feature. There may be autonomic involvement and respiratory compromise.
A strategically placed lesion involving the pyramidal decussation may cause unusual patterns of weakness. The corticospinal fibers innervating the upper extremities are thought to decussate more rostrally and medially than the fibers innervating the lower extremities, although this concept has been questioned (Figure 11.12). The term cruciate paralysis is used in two ways. One refers to weakness of both arms, brachial diplegia, with relative sparing of the legs, due to a lesion involving the rostral portion of the pyramidal decussation. The findings are similar to those of a central cord syndrome of the cervical spine or the man-in-the-barrel syndrome due to watershed cerebral infarction. Most cases are due to trauma. The other use refers to corticospinal paralysis of one arm and the opposite leg (cruciate hemiplegia, pyramidal decussation syndrome). This may occur because a lesion involves arm fibers that have already decussated but leg fibers that have not, which causes a crossed pattern of weakness. Triparesis, with weakness of one arm and both legs, has been reported after unilateral medial medullary infarction.
Gerstman-Straussler-Schinker (GSS) syndrome is a rare autosomal dominant spongiform encephalopathy due to a mutation of the prion protein gene. It begins in midlife and runs a progressive course with ataxia, spasticity, dysarthria, nystagmus, and dementia. GSS is genetically and phenotypically heterogeneous; among the different prion diseases, it has the longest clinical course and the potential to mimic other neurologic disorders, such as cerebellar degeneration and demyelinating disease.
Basilar artery (Bickerstaff’s, basilar type, vertebrobasilar, posterior fossa) migraine is an unusual type of complicated migraine with prominent brainstem symptoms similar to those of VBI. The disorder occurs primarily in young females and is usually followed by an occipital headache.
The foramen magnum syndrome can cause some unusual and puzzling clinical deficits. Lesions in the region of the foramen magnum are typically compressive extramedullary mass lesions (e.g., meningioma). Patients may have crossed hemiparesis, involving one arm and the opposite leg, because of involvement of the pyramidal decussation (see above). There may be weakness and wasting of the small hand muscles for reasons that remain unclear. Such hand muscle wasting may also occur as a false localizing sign in upper cervical spinal cord compression. Downbeat nystagmus in primary gaze is suggestive of a lesion at the cervicomedullary junction, and the nystagmus is often greatest in eccentric downgaze. Other symptoms suggestive of a foramen magnum lesion include occipital headache, neck pain, and stiffness; Lhermitte’s sign; C2 sensory loss; and shawl distribution upper extremity sensory loss. Tumors are generally histologically benign and often become large before the diagnosis is made. Masses usually intrude from posteriorly, so that posterior column signs, including pseudoathetosis, are common. Lower CN palsies are uncommon. There may be a fluctuating course simulating MS.
Bulbar Palsy
There are two principal types of bulbar palsy: PBP and pseudobulbar palsy. In both, the outstanding symptoms are dysphagia and dysarthria; both run a chronic course. Despite the similarities, the etiologies are different.
PBP is a form of motor neuron disease in which the disease attacks bulbar innervated muscles. There is weakness and atrophy of muscles supplied by the lower CNs, often accompanied by fasciculations. It is closely related to progressive spinal muscular atrophy, in which the process is limited to the anterior horn cells of the spinal cord, and amyotrophic lateral sclerosis (ALS), in which there is involvement of the bulbar nuclei, the anterior horn cells, and the pyramidal cells in the motor cortex.
In PBP, there is a relentlessly progressive degeneration of the neurons of the brainstem motor nuclei, primarily those in the medulla. It usually occurs in late adult life with onset in the sixth and seventh decades. The disease usually starts in the nucleus of the CN XII and ascends. Typical initial manifestations are atrophy, weakness, and fasciculations of the tongue. Involvement is bilateral from the outset. In advanced cases, the patient may be unable to protrude the tongue or to manipulate food in the mouth. The lingual involvement is followed or accompanied by dysphagia, usually for both liquids and solids, and by dysarthria. Nasal regurgitation of liquids is common and may lead to choking and aspiration. Involvement of the soft palate, larynx, and tongue causes flaccid dysarthria. The speech is “thick,” as though the mouth were filled with soft food, with a nasal component. Early, the most pronounced difficulty is with pronunciation of linguals and velars; later, the labials are affected. In advanced cases, speech is reduced to unintelligible laryngeal noises. There is often marked drooling of saliva. Patients may keep a tissue or rag at the chin to absorb unswallowed secretions. Sometimes atrophy and fasciculations extend to the palate and pharynx, and the condition may eventually ascend to involve the facial and trigeminal motor nuclei. Occasionally, the sternocleidomastoid and trapezius muscles are affected. There may be autonomic involvement with tachycardia. The palatal and pharyngeal gag reflexes disappear early. There are no sensory changes. PBP is aggressive and relentless, with death usually caused by aspiration pneumonia. PBP may be the first manifestation of ALS. When ALS causes prominent bulbar weakness, it is referred to as bulbar ALS. In bulbar palsy due to ALS, there are also corticospinal tract manifestations. In a series of 32 patients with PBP, all but two progressed to ALS, regardless of the presence of upper motor signs or generalized denervation on limb EMG. The other two died at the PBP stage.
Severe bulbar involvement occurs in other motor neuronopathies. It is often the terminal aspect of Werdnig-Hoffmann disease (hereditary spinal muscular atrophy type 1). Fazio-Londe disease is PBP occurring in children. Kennedy’s disease (X-linked recessive bulbospinal neuronopathy) causes a clinical picture resembling ALS but with slow progression and other atypical features; dysphagia or dysarthria may be prominent late in the course. Bulbar polioencephalitis may occur as part of paralytic poliomyelitis, causing paralysis of the throat, tongue, and respiratory muscles. Creutzfeldt-Jakob disease may present as bulbar palsy.
In pseudobulbar palsy, there is also marked difficulty with bulbar function, including speech and swallowing. Although the clinical manifestations are similar, the underlying mechanism is entirely different. Pseudobulbar palsy is caused by bilateral supranuclear lesions, which involve the corticobulbar pathways to the bulbar nuclei. PBP and bulbar ALS cause lower motor neuron weakness; pseudobulbar palsy causes upper motor neuron weakness. In patients with bulbar ALS, both processes may be at work. Because of bilateral supranuclear innervation, unilateral lesions of the corticobulbar tract rarely cause significant bulbar dysfunction. But with bilateral supranuclear lesions, the bulbar dysfunction may be severe. It is usually accompanied by other upper motor neuron signs. There may be weakness and spasticity of the muscles of mastication, an exaggerated jaw jerk, and frontal release signs such as snout and suck reflexes. Difficulty with emotional control causing spontaneous, unprovoked laughing and crying (emotional incontinence) is common. Pathologic laughing (crazy laughter or “fou rire prodromique”) and crying have also been reported with brainstem lesions. Some patients have paresis of the muscles of facial expression causing masking of the facies. There are typically significant neurologic abnormalities beyond the distribution of the CN nuclei, with bilateral cortical spinal tract signs.
The most common cause of pseudobulbar palsy is multiple cerebral infarctions. The syndrome may also occur in encephalitis, MS, trauma, cerebral anoxia, primary lateral sclerosis, or other disease processes that cause bilateral corticobulbar tract lesions. The lesions may be in the cortex or in the corona radiata, internal capsule, cerebral peduncles, or brainstem rostral to the nuclear centers. Speech is thick and slurred but may have an explosive quality. There may be dysphagia, nasal regurgitation, choking, and drooling. Patients may keep food in the mouth for prolonged periods. There is less of a tendency to choke than in true bulbar palsy because the gag reflexes are intact and may be hyperactive. Although the tongue may be strikingly immobile, atrophy and fasciculations do not develop. The prognosis in pseudobulbar palsy is no more favorable than in PBP. The eventual outcome in both conditions is death, often because of aspiration. Two types of pseudobulbar palsy have been described; one is due to lesions affecting the corticobulbar fibers, and the other is due to involvement of the basal ganglia or extrapyramidal pathways. In striatal pseudobulbar palsy, there are additional signs of basal ganglia involvement, including rigidity, hyperkinesias, and a parkinsonian picture.
Other conditions that may cause prominent weakness of bulbar muscles or other evidence of brainstem dysfunction include neuromuscular transmission disorders, some neuropathies and myopathies, and certain rare neurologic conditions. The dysarthria and dysphagia of myasthenia gravis (MG) may resemble bulbar palsy. Early in the course, it may be difficult to distinguish bulbar ALS or PBP from MG. The characteristic eye signs of MG are not always present. Bulbar signs and symptoms similar to those of MG can occur in botulism and Lambert-Eaton syndrome. Bulbar muscle weakness can occur in muscular dystrophies, especially oculopharyngeal dystrophy, and other myopathies. Bulbar weakness may complicate Guillain-Barré syndrome and other polyneuropathies. CN involvement is characteristic of diphtheritic polyneuropathy, causing a bulbar syndrome with dysarthria and dysphagia due to weakness of the soft palate, pharynx, and tongue. In tetanus, pharyngeal spasms may accompany trismus. In rabies, spasmodic contractions of the muscles occur on attempts to swallow. Whipple’s disease involving the central nervous system (CNS) may have prominent brainstem findings. Oculomasticatory myorhythmia, a striking movement disorder involving the eyes and jaw, is characteristic, perhaps pathognomonic, of CNS Whipple’s disease. Brainstem involvement may be a striking feature of Leigh’s disease (subacute necrotizing encephalomyopathy). The brainstem can also be damaged by radiation.
MULTIPLE CRANIAL NERVE PALSIES
Intracranial-extramedullary or extracranial processes may involve more than one CN. A disease may involve homologous nerves on the two sides (e.g., bilateral facial palsy) or different nerves on the same or opposite sides. In some conditions, a cluster of nerves is involved in a discrete anatomical region. The progression may follow some anatomical pattern or appear capricious. Multiple CNs may be affected from the outset, or the process may begin with one nerve and progress to involve others. Pain may or may not be present. Table 21.3 lists some conditions that may cause MCNPs. Table 21.4 covers some of the named multiple CN syndromes. The patient shown at http://www.youtube.com/watch?NR=1&v=S9s5ZHCzOXMX has multiple cranial neuropathies.
TABLE 21.3 Some Disease Processes that May Involve Multiple Cranial Nerves (CNs)

CMV, cytomegalovirus; EBV, Epstein-Barr virus; HTLV-1, human T-cell lymphocytotrophic virus.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


