Fig. 21.1
Histopathological changes in diffuse axonal injury: espheroids or retraction balls in injured axons in white matter. (a) hematoxylin eosin staining (×75). (b) immunohystochemistry for neurofilaments (68 KD). (c) antibodies against ubiquitin (×150) (Modified with permission from Jose Luis Palomo Rando. From Lafuente JV. Diffuse axonal injury. Diagnostic importance in forensic neuropathology. Cuadernos de Medicina Forense. 2005;11(41):173–82.) (3)
Impact forces generated by head trauma are of high magnitude and of relatively short duration (5–200 ms). They produce acceleration forces on the head and brain. During the impact, the head’s center moves along a straight line (translation) and around its center of gravity (rotation), being the last yet the most important. Macroscopic lesions such as fractures, hematomas, or edema are hardly ever seen (less than 1 %) (7). However, at a later time, that is, after several hours to days/weeks, microscopic and metabolic changes can be observed (Fig. 21.2). These changes underlie both biochemical events that cause neuronal damage and biomechanical events resulting in axonal injury (4).
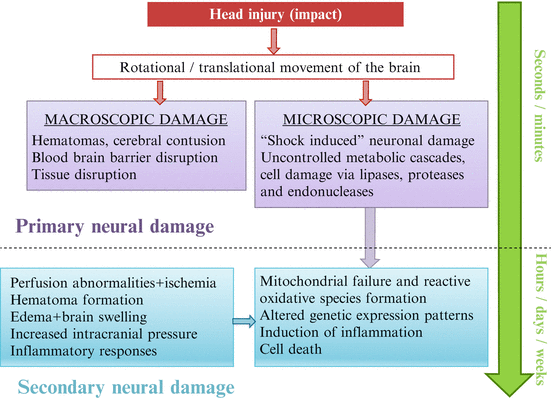
Fig. 21.2
Sequence of different mechanisms that lead to neural damage after traumatic brain injury (Adapted by permission from BMJ Publishing Group Ltd. Anderson T, Heitger M, Macleod AD. From Concussion and mild head injury. Pract Neurol. 2006;6:342–57) (1)
Biochemical Events
Neuronal damage and death associated with MTBI have been widely studied in the past decades (8,9). However, little has been documented about remote and diffuse neuronal changes after trauma (10,11). Studies have shown evidence of neuronal death, bilaterally, in cerebral sites remote from the impact, primarily within the neocortex, hippocampus (pyramidal layers C1, C2 and C3) (10, 11), and diencephalon, in addition to the striatum, both inferior and superior colliculli and the cerebellum. These cells show ultrastructural changes of both necrosis and apoptosis (10).
Despite evidence of neuronal death far from the impact site, the involved mechanisms are not understood. It is likely that other potentially important factors and mechanisms associated with direct mechanical perturbation and its sequelae may be at work in the pathogenesis of this diffuse necrotic neuronal death. As such, they should be taken into account (10).
At the present time, apoptotic cell death is believed to play a role in delayed neuronal death occurring several hours to weeks after diffuse traumatic brain injury (DTBI). The dominance of proapoptotic factors in the presence of a persistent energy supply results in the activation of cysteine proteases such as caspases that are regulators and effectors of apoptotic cell death (10). Changes include altered expression of DNA transcription factors and apoptotic proteins, uncontrolled release of proteases, lipases, and endonucleases, with the subsequent oxidative stress. Collectively, these changes cause degradation of cell membrane and cytoskeletal proteins, mitochondrial failure, and production of free radicals. Although both necrosis and apoptosis mechanisms have been well described and different pathways are known to take part in some non-traumatic central nervous system (CNS) pathologies, their specific role and pathways in TBI need still to be studied (10).
Neuronal death in localized areas leads to the release of cytokines and other proinflammatory molecules. This fact induces an inflammatory response, which is believed to be responsible for secondary brain injury. It increases permeability of the blood brain barrier and activates microglia. Enough is not known about these changes occurring after minor injury. There is some evidence suggesting that cellular signaling after neuronal injury and the resultant cytokine cascade, microglial response and cytopathological alterations might lead to amyloidogenesis and accumulation of amyloid beta-peptide (A-beta). A-beta is toxic to neuroglia and plays a major role in neurodegenerative changes following some types of brain injuries such as trauma and stroke. Indeed, damaged axons can become a reservoir for A-beta, contributing to A-beta plaque formation after MTBI. These quiet changes can add up after several concussions and produce brain dysfunction (12). This could for instance, explain cognitive impairment and dementia seen after repeated sports concussion.
Alterations in neurotransmission, both in excitatory and inhibitory systems, may also play a role in long-term deficits in memory and cognition. Among others, long-potentiation, an N-methyl-D-aspartate (NMDA)-dependent measure of plasticity seems to be persistently impaired in the hippocampus after concussion. It also produces changes in choline acetyltranspherase activity and loss of cholinergic neurons in the forebrain. In addition, there is a loss of GABAergic neurons, which can compromise normal inhibition of hippocampal dentate granule cells, facilitating seizures (2).
Finally, as seen with different animal models of brain injury, blood brain barrier (BBB) and its disruption play an important role in secondary neural damage. Deep changes in BBB permeability have been seen around areas of micronecrosis, partially mediated by the release of vascular endothelial growth factor ( 13, 14). These alterations may accompany other mechanisms occurring after MTBI.
Biomechanical Events
After a brief review of neurochemical cascades triggered by the forces of injury, we shall consider the mechanically induced neuronal death cascades led by direct perturbation of the neuronal cell membrane or its prolongations.
Although axotomy-related neuronal somatic changes occurring adjacent to sites of traumatic axonal injury had been previously recognized, the relation to any subsequent injury cascades has not yet been appreciated. A recent study using immunohistochemical techniques to recognize amyloid precursor protein (APP), a well-accepted marker of traumatic axonal injury (TAI), identified some neuronal somata directly linked to it in the cerebral cortex, hippocampus, and thalamus. It showed that despite contemporary thought, even in immediate proximity to the neuronal soma, TAI did not result in acute neuronal death. Instead, those neurons revealed reactive changes. These changes included loss and degranulation of the rough endoplasmic reticulum, disaggregation of polysomes, and dispersal of the Golgi apparatus, as well as transient suppression of protein synthesis. Furthermore, there was no cytoskeletal or mitochondrial alteration. In combination these transient reactive changes suggest a potential neuronal attempt at reorganization and repair, rather than the initiation of any prenecrotic/apoptotic change.
Using extracellular tracer infusion techniques, multiple in vitro and in vivo studies have demonstrated that immediately following DTBI, non-axotomized neurons can take up high molecular weight tracers. For example, horseradish peroxidase or other molecular weight dextrans and other molecules normally excluded from the neuronal cytoplasm by the intact cell membrane, are taken up. This immediate tracer uptake suggests that the mechanical force of the injury itself evoked neuronal cell membrane disruption (mechanoporation). It is probable that this phenomenon allowed the influx of damaging ions through the compromised cell membrane (11).
Following these initial descriptions, subsequent studies revealed a rather heterogeneous neuronal response to axonal disruption. Some of them showed ultrastructural signs of necrosis while others did not. Furthermore, at the same time and in the same brain foci, different populations of injured neurons did not reveal evidence of membrane disruption. Instead, some of them showed perisomatic axonal injury resulting in increased neuronal somatic APP-positivity. Others exhibited non-axotomy-related induction of heat shock protein expression. After this, the potential resealing of the neuronal membrane leading to its recovery became a possibility to be taken into account (11). By administrating different extracellular tracers at varied times post-injury, in vitro studies revealed that the majority of neurons sustaining mechanoporation could reseal their disrupted membranes in the first few minutes post-TBI. However, in vivo studies did not confirm these observations. Rather, the results suggested that independent of mechanoporation resealing, areas of enduring membrane permeability were present. To further complicate things, recent findings show not only occurrence of potentially delayed membrane resealing, but also of the delayed membrane disruption (10). To date, there are no data that support neuronal death caused only by mechanical disruption.
Although the underlying mechanisms of this delayed membrane disruption are unknown, a sustained elevated intracranial pressure persisting for several hours after TBI could probably contribute to this phenomenon.
In addition to these changes, diffuse axonal injury has also been a distinguishing feature of traumatic head injury, even MTBI, for some decades (3,8). Contemporary studies have shown that in large part, the traditionally accepted premise of transection and retraction of the axon is not correct. Rather, it has been seen that the forces of injury diffusely alter focal axonal segments. This results in a local impairment of axonal transport, with progressive local axonal swelling followed by detachment over a post-traumatic course ranging from several hours up to a day. Given the fact that these reactive axonal changes were found in scattered axons related to other intact axons, this precluded the potential for direct mechanical renting. It suggested that more subtle intraaxonal changes were at work in the pathogenesis of progressive axonal disconnection.
Emphasis has been directed on identifying the initiating intraaxonal cellular and subcellular factors for a better understanding of pathobiology of axonal injury and to develop treatment therapies. While immediate physical transection of the axon cylinder has been ruled out, the potential for focal disturbances in the axolemma leading to local ionic dysregulation was proposed. It was then demonstrated that damage in the mitochondria released cytochrome C. In turn, caspase-mediated spectrin degradation is activated (13). Additionally, degradation of the axonal cytoskeleton associated with concomitant neurofilament side-arm cleavage, neurofilament compaction, and microtubular loss were activated. Therefore, it is generally assumed that these intraaxonal mechanisms lead to an upstream impairment of axonal transport, which results in swelling and disconnection. However, additional recent studies trying to find spatial relation between transport impairment and axonal permeability disturbances have shown no correlation (14).
Therefore, all this research evidence shows the neurofilament-compacted axonal segments and swollen axonal segments demonstrating impaired axonal transport. It suggests that current findings are most likely evidencing the existence of two different populations of injured axons. These populations would respond differently to the traumatic episode. For those segments showing focally altered axolemmal permeability it appears that local calcium dysregulation activates cystine protease and causes local degradation of the axonal cytoskeleton (14). The reason why these sites of axonal injury do not reveal impaired axonal transport and axonal swelling is still unclear. It is conceivable that the suprathreshold calcium uptake occurring at these sites most likely converts anterograde to retrograde transport. Thereby it precludes the development of reactive axonal swelling. This theory is supported by the use of various therapeutic strategies targeting calpain inhibition. Thus, calpain inhibition significantly reduces the number of axonal profiles showing the above described cysteine protease activation and cytoskeletal collapse (15).
In contrast, it appears that those axons showing impaired axonal transport and local swelling do not sustain any alteration of those described previously. Rather, it is posited that other mechanisms are at work and that these are linked to more subtle forms of calcium dysregulation. Activation of micromolar calpains which trigger the activation of calcineurin may be involved. In turn, calcineurin alters the microtubular network to disrupt local axonal transport kinetics and thereby elicits the swellings described above (16). Limited direct evidence exists to support this pathway. However, the use of calcineurin antagonists such as FK506 directly attenuates the number of axons showing impaired axonal transport and swelling. Moreover, these antagonists have no effect on those axons showing neurofilament compaction and disconnection. This supports the premise that calcineurin is integral to the pathogenesis of impaired axonal transport and swelling.
Collectively, these studies illustrate the complexity of the pathogenesis of diffuse axonal injury, suggesting at least two differing types of initiating mechanisms. There is the caveat that both populations of injured axons will likely not be amenable to one form of therapeutic intervention.
Finally, other mechanisms that might participate in DTBI are being studied. All the changes we have discussed above take into consideration only the myelinated axons. Unmyelinated axons have not received the same attention in the studied CNS pathology. Recently, however, electrophysiological studies have demonstrated damage and dysfunction in unmyelinated nerve fibers within the corpus callosum of traumatically brain-injured animals. They have shown significant and sustained depression of the compound action potentials (17). It has been seen in microscopical studies that in vitro, non-myelinated neurites subjected to mechanical strains ultimately led to local axonal swelling and disconnection. It occurred without any evidence of axolemmal change or disruption. Rather, the depolarization associated with this injury evoked sodium influx, the activation of voltage gated calcium channels and the concomitant activation of sodium/calcium exchangers, all of which contributed to local intraaxonal calcium overloading. These calcium-mediated changes were linked with the activation of proteases, which in turn contributed to subsequent proteolysis of its NaCh subunit, promoting a persistent elevation in intracellular calcium. This finally resulted in pathological changes through many of the pathways described before. Although this remains to be confirmed in vivo, it would involve a potential channelopathy as major player in the ensuing unmyelinated axonal perturbation.
Additional evidence suggests that altered dendrite structure may underlie the cognitive deficits observed after MTBI. At the cellular level, changes in dendrite structural proteins such as microtubule-associated protein 2 (MAP2) and neurofilament proteins are present in animal models and in autopsy specimens. Some studies show that stretch-induced axonal injury causes transient dendritic swelling, which was sodium-dependent, exacerbated by extracellular calcium removal and blocked by NMDA receptor antagonists. This finding may help us understand the mechanisms of cognitive symptoms after MTBI (18).
Related posts:
 Towards a Psychotherapy with a Philosophical Basis
Neurosciences, Neuroeconomics, and Metaphysics
Role of the Neuropeptide Angiotensin II in Stress and Related Disorders
Animal Models of Psychopathology and Its Relation to Clinical Practice
Adenosine in the Neurobiology of Schizophrenia: Potential Adenosine Receptor-Based Pharmacotherapy
Towards a Psychotherapy with a Philosophical Basis
Neurosciences, Neuroeconomics, and Metaphysics
Role of the Neuropeptide Angiotensin II in Stress and Related Disorders
Animal Models of Psychopathology and Its Relation to Clinical Practice
Adenosine in the Neurobiology of Schizophrenia: Potential Adenosine Receptor-Based Pharmacotherapy
 Translating the Glutamatergic Hypothesis of Schizophrenia Through Homeostatic Regulation of Brain Glycine
Translating the Glutamatergic Hypothesis of Schizophrenia Through Homeostatic Regulation of Brain Glycine
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


