3 Pathology and Radiology A. Neural stains—hematoxylin and eosin stain (H&E) for general pathologic examination, the Nissl stain for neuron cell bodies (binds nucleic acid), and the silver stain for cell processes. Lipofuscin accumulates in central nervous system (CNS) neurons with aging. Neuromelanin accumulates in neurons (especially the substantia nigra [SN] and locus ceruleus) and is a catecholamine waste product (it is not made by tyrosinase). True melanin is made by tyrosinase and is located in the leptomeningeal melanocytes of the ventral medulla and cervical cord. These cells form the primary CNS melanomas. B. Edema (increased brain water) 1. Vasogenic edema—due to increased blood–brain barrier (BBB) permeability to proteins and macromolecules. It is caused by vessel damage, inflammation, and neovascularity. It is the most common type of edema, is extracellular, and affects the white matter more than the gray matter. Corticosteroids decrease the edema. 2. Cytotoxic edema—caused by an impaired Na+/K+ pump caused by decreased adenosine triphosphate (ATP) delivery during hypoxia and ischemia. Water and electrolytes accumulate in the cell, but not plasma proteins. It is intracellular, affects both the gray and white matter, and is not associated with computed tomographic (CT) or magnetic resonance imaging (MRI) enhancement because of the intact BBB. Steroids do not decrease the edema. 3. Interstitial edema—the transependymal shift of fluid caused by cerebrospinal fluid (CSF) accumulation (hydrocephalus) and is extracellular C. Lethal neuronal injury—characterized by ischemic necrosis. The cytoplasm that was slightly basophilic becomes eosinophilic (red is dead) and the nucleus becomes shrunken and dark. Changes can be seen within 6 hours. Ferrugination occurs when dead neurons become encrusted with Fe2+ and Ca2+ salts. D. Progressive or reversible neuronal injury is characterized by 1. Central chromatolysis—occurs after an injury to an axon near the cell body. The Nissl substance disappears, the nucleus becomes eccentric, and the cell body enlarges. It is seen in the anterior horn cells with anterior nerve root compression and Guillain–Barré syndrome. The cells may progress to death or recover. 2. Neurofibrillary tangles—Argyrophilic linear densities accumulate in the cell bodies and processes. They can be detected on silver stain. The neurofibrils are made up of hyperphosphorylated tau proteins, neurofilaments, and microfilaments (actin). They are much more numerous with Alzheimer disease, postencephalitic Parkinson disease, progressive supranuclear palsy, and aluminum toxicity. 3. Neuronal storage of lipids or carbohydrates—The cell body enlarges, the nucleus becomes eccentric, and the cytoplasm becomes foamy. a. Viral (1) Intranuclear—herpes simplex virus 1 (HSV-1; Cowdry type A, eosinophilic, in neurons, astrocytes, and oligodendrocytes; seen early in the disease), cytomegalovirus (CMV), and measles (subacute sclerosing panencephalitis [SSPE]) (2) Intracytoplasmic—rabies (Negri bodies), SSPE, and CMV b. Degenerative/metabolic (all intracytoplasmic)—Pick bodies (in Pick disease, round stain with silver), Lewy bodies (in Parkinson disease, have a halo), Lafora bodies (in Lafora disease, periodic acid Schiff [PAS] positive, basophilic, have a dense core), Hirano bodies (eosinophilic, most in hippocampus, made of actin, seen in the elderly and increased number with Alzheimer disease), and Bunina bodies (eosinophilic, increased with amyotrophic lateral sclerosis [ALS]) 5. Marinesco bodies are seen in normal brain. They are eosinophilic and contain chiefly ubiquitin. They are located mainly in the nucleus of melanin cells of the brainstem. E. Neuronal atrophy—occurs with degenerative diseases and has been postulated to be due to either excitatory amino acids, subcellular injuries by free radicals, or gene-directed apoptosis. Neuronal atrophy is associated with proliferation of astrocytes and microglia. F. Hamartoma—disorganized cells in the proper location for that cell type G. Choristoma—correctly organized cells in the wrong location H. Brain herniation changes—Kernohan notch (the contralateral cerebral peduncle is compressed against the incisura with ipsilateral weakness producing a false localizing sign), posterior cerebral artery (PCA) stroke, Duret hemorrhages of the midbrain and pons (by arteriole stretching), CN III palsy, and hydrocephalus (by compression of the cerebral aqueduct). A. Astrocytes—either protoplasmic (mainly in the gray matter) or fibrillary (mainly in the white matter). Glial fibrillary acidic protein (GFAP) is the main component of astrocytic intermediate filaments. B. Astrocytic reaction to injury 1. Secondary (reactive) astrocytosis—occurs after stroke, degenerative diseases, etc. There rarely are mitotic figures unless there is neoplastic disease. Gemistocytic astrocytes are large reactive astrocytes with eccentric nuclei. Fibrillary astrocytes appear later, have smaller cell bodies, and more fibers. Rosenthal fibers are eosinophilic masses in the astrocytic processes and are increased with Alexander disease, pilocytic astrocytomas, and reactive astrocytosis. 2. Primary astrocytosis—the proliferation of astrocytes after astrocytic disease such as hepatic encephalopathy with Alzheimer II astrocytes (large nuclei, gray, glycogen inclusions) (Fig. 3.1) C. Microglia—macrophages from outside the CNS. With mild injury, there may be rod cells with cigar-shaped nuclei. D. Oligodendrocytes—proliferate around multiple sclerosis plaques and are increased in size with bizarre shapes around the periphery in progressive multifocal leukoencephalopathy (PML) E. Ependymal cells—do not proliferate with injury but when damaged are replaced by subependymal astrocytes Fig. 3.1 Alzheimer type II astrocytes (hematoxylin and eosin [H&E] stain) with large vesicular nuclei and cytoplasm (arrows). A. Conventional x-rays—usage largely supplanted by the widespread availability and rapidity of CT scans. In the pediatric population skull x-rays continue to have a defined role in the evaluation of minor head trauma and suspected child abuse, metallic foreign objects implanted in the skull, abnormal head shape, and integrity of ventricular shunt tubing as part of a “shunt series.” Spinal x-rays continue to be performed in the preliminary evaluation of spinal trauma and to assess spinal stability. B. Multislice CT scans—x-ray-based method remains investigation of choice for preliminary radiographic evaluation of the CNS. Particularly well suited for identifying bony lesions and acute hemorrhage, for example, in the evaluation of trauma and stroke. The ability of modern scanners to perform 3D-reconstruction is particularly advantageous in the evaluation of traumatic spinal injuries. 1. CT angiography (CTA)—involves administration of iodinated contrast agent; well-suited for evaluation of carotid and vertebral atheromatous disease and dissection, cerebral aneurysms, and acute thrombi within intracranial vessels 2. CT perfusion (CTP)—involves administration of iodinated contrast agent and can be rapidly performed on any standard helical CT scanner. Its main usage is distinguishing between “at risk” and infarcted brain in stroke patients. The following perfusion parameters are typically measured: cerebral blood volume, cerebral blood flow (CBF), mean transit time, and time to peak enhancement. C. MRI—the modality of choice for resolution of anatomic detail and subtle disease processes. Unlike CT scans, it does not use ionizing radiation. MRI is based on electromagnetic fields generated by hydrogen nuclei. Prior to the application of an external magnetic field, the magnetic axes of hydrogen nuclei are randomly aligned. Upon exposure to an external magnetic field, these axes are forced into alignment with the external source to produce a net magnetization vector. When a radiofrequency (RF) pulse is then applied, the net magnetization vector tilts to produce a vector with two constituent components—a longitudinal vector and a transverse vector. The transverse vector contributes to the return RF pulse, or “echo,” to generate the MR signal. When the RF pulse is turned off, the net magnetization vector realigns with the external source. This occurs through two simultaneous processes: T1 recovery and T2 decay. T1 recovery is the time constant for the “recovery” of magnetization along the longitudinal axis. T2 decay reflects the concomitant decrease in magnetization along the transverse axis. Different tissues have different T1 and T2 values, which serve as the basis for tissue contrast in all MR imaging. Moreover, tissue contrast may be preferentially dependent on either T1 recovery or T2 decay, and images can be generated that are relatively T1fig or T2-weighted to maximize these contrasts. 2. Fluid-attenuated inversion-recovery (FLAIR)—special spin-echo sequence that eliminates signal from CSF. FLAIR MRI is most useful in the delineation of cystic lesions and tumors abutting sulci or the ventricles. 3. Gradient echo (GRE) sequences—extremely fast sequence that is particularly sensitive to magnetic field inhomogeneity that results in a local loss of signal. This sequence is well suited for the detection of hemorrhage, e.g., cerebral contusions, cavernous malformations, where the iron in hemoglobin causes its own magnetic field. GRE sequences form the basis of MR perfusion studies (which require the administration of a gadolinium-based contrast agent) and functional MRI (fMRI, discussed below). 4. Diffusion-weighted imaging (DWI)—distinguishes rapid (unrestricted diffusion) and slow diffusion of protons (restricted diffusion). When coupled with apparent diffusion coeffcient (ADC) maps, this technique allows determination of stroke age. Acute stroke will appear bright on DWI and dark on ADC maps. The reverse is true for chronic stroke. 5. MR angiography (MRA)—useful for visualization of the cerebral vasculature a. Time-of-flight (TOF)—a rapid sequence that does not require intravenous (IV) administration of a contrast agent. The technique involves suppression of MR signal from stationary tissues so that flowing blood appears relatively hyperintense. b. Contrast-enhanced MRA—requires IV administration of a gadolinium-based contrast agent. The paramagnetic contrast agent serves to shorten the T1 of blood, which results in high signal intensity on T1-weighted MRIs. 6. MR spectroscopy (MRS)—a water suppression technique that permits the detection of brain metabolites. MRS can be performed at the same time as conventional MRI with appropriate software. Clinically, it is most useful in the differentiation of tumor from nonneoplastic processes such as abscess, tumefactive demyelination, and subacute infarcts. Brain tumors typically demonstrate reduced N-acetylaspartate (NAA) and creatine levels and increased choline levels compared with normal brain. 7. fMRI—used for functional mapping of the brain, in particular, preoperative mapping of eloquent cortex (e.g., primary motor, sensory, and language cortex). The underlying premise of fMRI is the observation that increased neuronal activity is associated with increased CBF and oxygen delivery out-of-proportion to the tissue’s capacity to consume the increased oxygen. The result is a local increase in the concentration of intravascular oxyhemoglobin (nonparamagnetic substance) and a concordant decrease in the concentration of deoxyhemoglobin (paramagnetic substance). The locally decreased concentration of a paramagnetic substance allows for less severe local magnetic field distortions and an increased MRI signal. fMRI does not require IV administration of a contrast agent. D. Digital subtraction angiography (DSA)—prior to CT scans and MRI, DSA (an x-ray-based technique that involves the administration of iodinated contrast agent) was the test-of-choice in the evaluation of suspected intracranial tumors whose mass effect caused the displacement of cerebral vascular structures. Today DSA remains the gold standard in the evaluation of cranial and spinal vascular disease. Therapeutic neurointer ventional radiology involves the deployment of coils, embolic materials, and stents for the treatment of aneurysms, vascular malformations (arteriovenous malformations [AVMs], dural arteriovenous fistulas), and carotid atheromatous disease. Preoperative tumor embolization is sometimes a useful surgical adjunct in the treatment of meningiomas. 1. Brain tumors—increased tumor metabolic rate, as measured by the uptake of a glucose analog, 18F-2-deoxy-D-glucose, may help differentiate low- versus high-grade gliomas, tumor recurrence versus radiation necrosis and primary CNS lymphoma versus toxoplasmosis in acquired immunodeficiency syndrome (AIDS) patients. It may also be useful in monitoring subclinical response to radiation and chemotherapy and assessing for malignant transformation of low-grade lesions. Other radioactive tracers, e.g., radiolabeled amino acids, have shown early promise in the evaluation of brain tumors. 2. Epilepsy—clinical utility generally restricted to patients with focal epilepsy being evaluated for surgical therapy. Interictal PET demonstrates lesional reduced glucose metabolism and CBF. An opposite pattern is seen during an ictal event. Interictal PET may also be useful in localization of eloquent cortex. 3. Emerging areas—early diagnosis of Alzheimer disease and other dementia syndromes, diagnosis and differentiation of Parkinsonian syndromes, evaluation of “at-risk brain” in patients with cerebral ischemia A. 3% of newborns have major structural abnormalities, and most involve the CNS. B. The brain and spinal cord are formed from neuroectoderm at 3 to 8 weeks. Primary neurulation occurs at 3 to 4 weeks with formation of the neural plate, neural groove, and neural folds. The primitive streak forms at postovulatory day 13. The notochord forms at day 17 and induces the primitive streak to form the neural plate that later develops the neural groove and neural folds. The neural folds fuse at 22 days to form the neural tube, with the proximal two thirds forming the brain and the distal one third forming the spinal cord. It closes like a zipper starting at the hindbrain with the anterior neuropore closing first at 24 days (forming the lamina terminalis) and the posterior end closing second (to L1/2) at 26 days. A problem at this stage causes neural tube defects and Chiari malformations. C. Dysjunction—the separation of ectoderm from neuroectoderm after the neural tube forms. The mesenchyme in between the two layers forms dura, neural arches, and paraspinal muscles. If dysjunction occurs too early, the mesenchyme can enter the neural tube and form lipomas and lipomyelomeningoceles. Focal failure of dysjunction causes an epithelial-lined dermal sinus tract. More widespread failure of dysjunction may cause a myelocele or a myelomeningocele. D. Secondary neurulation—occurs at 4–5 weeks as the mesoderm forms the dura, skull, vertebrae, and distal spine. Defects of secondary neurulation cause spinal dysraphism below L1/2. F. Neuronal proliferation and differentiation—occur at 2–4 months. A problem at this stage causes vascular malformations and neurocutaneous syndromes. G. Cellular migration—occurs at 2–5 months with migration of cells from the deeper layers to more superficial layers, except for the outer cortical layer. A problem at this stage causes callosal agenesis, schizencephaly, and heterotopias. H. Neuronal organization and myelination—occur from 5 months to the postnatal period as the synapses form. Myelination begins in the fifth fetal month and proceeds caudad to cephalad, dorsal to ventral, central to peripheral, and sensory before motor. Most is completed by 2 years. At birth, the cortex/white matter signals on MRI are reversed because of the paucity of myelination and increased water content of the cortex compared with the white matter. I. At 4–5 weeks, the prosencephalon, mesencephalon, and rhombencephalon form. The prosencephalon then divides into (1) the telencephalon that forms the hemispheres, caudate, putamen, fornices, anterior commissure, corpus callosum, and hippocampus; and (2) the diencephalon that forms the thalamus, globus pallidus (GP), posterior hypophysis, infundibulum, optic nerve, retina, posterior commissure, and habenular commis-sure. The mesencephalon forms the midbrain. The rhombencephalon divides into (1) the metencephalon that forms the pons, fourth ventricle, and cerebellum; and (2) the myelencephalon that forms the medulla. J. The germinal matrix—forms at 7 weeks and produces neurons and glia. At 30 weeks, the germinal matrix involutes (although some clusters exist until 39 weeks). K. The commissures—form at 8–17 weeks. The corpus callosum forms from front to back except for the rostrum that forms last, so partial agenesis always includes the rostrum and splenium. L. Cells proliferate in the walls of the neural tube and form a pseudostratified columnar layer with ventricular and subventricular zones (precursors of neurons and glia). Processes of these cells go from the lumen to the external basement membrane forming the marginal zone with an underlying intermediate zone. Most cells proliferate in the ventricular zone and migrate outward by ameboid movement guided by glial processes that extend from the ventricular zone to the pial surface of the marginal zone. The migration is dictated by intercellular adhesion molecules and integrins (cell surface receptors). The cells mature, selectively die, group together, and form connections. M. Brainstem—forms between 2–6 months. The basal plate has (medial to lateral) somatic efferent, special visceral efferent, and general visceral efferent nuclei. The alar plate has (medial to lateral in the plate and lateral to medial in the final position) general visceral afferent, special visceral afferent, and somatic afferent nuclei. Both efferent and afferent somatic nuclei are the most medial of their groups in the full-term human. N. Spinal cord—has a sulcus limitans that forms between the dorsal (alar) and ventral (basal) plates. The connections form at 2–4 months. O. Caudal spinal cord—forms separately by retrograde differentiation at 4–8 weeks as the caudal cell mass forms and cavitates. P. Neural crest—the group of cells at the neural tube/somatic ectoderm junction. It forms the leptomeninges, Schwann cells, sensory ganglia of the cranial nerves, dorsal root ganglia (DRG), autonomic nervous system ganglia, the adrenal medulla, melanocytes, and amine precursor uptake and decarboxylation (APUD) cells. R. Notochord remnants—form the nucleus pulposus of the intervertebral disks. They are also believed to be the cells of origin of chordomas. S. Dura—formed from mesodermal elements, and the pia/arachnoid is formed from neuroectoderm T. Full-term brain—weighs 400 g and is 90% water (the adult brain is 70% water). It has larger nuclei with less arborization. The anterior and lateral corticospinal tracts are incompletely myelinated. The corpus callosum and the fornix are completely unmyelinated. There may be myelination glia where immature oligodendrocytes in the site of myelination may appear like reactive astrocytes. There frequently is a cavum septum pellucidum. The immature cells around the lateral ventricles and the external granular layer of the cerebellum are gone after 12–15 months. U. Suture closure—Metopic suture closes around the first year of life. V. The anterior fontanelle closes by 2.5 years of life. The posterior and sphenoid fontanelles close by 2–3 months of life. The mastoid fontanelle closes by 1 year of life. A. Malformation—occurs when an organ is not formed properly. Dysplasia occurs when a tissue is not formed properly. Developmental injury may be caused by genetic abnormalities, chromosomal aberrations, or environmental factors (infections, drugs, chemicals, malnutrition, and maternal illness). B. Neural tube defects (dysraphism)—caused by failure of fusion of the neural tube. 1. During neurulation (occurs at 17–30 days)—failure of fusion produces open defects by failure of dys-junction of the neural and cutaneous ectoderm. The risk is increased with a neural tube defect in a sibling or maternal folate deficiency. Maternal serum α−fetoprotein (AFP) usually is increased. This group includes spina bifida aperta, meningomyelocele, myelocele, anencephaly, and cranioschisis. 2. Postneurulation (occurs at 26–60 days or even near birth)—Failure of fusion produces closed, skin-covered deficits such as spina bifida occulta, holoprosencephaly, encephaloceles, hydrocephalus, diastematomyelia, lipoma, meningocele, and Chiari malformations (Fig. 3.2). 3. Anencephaly—the most common congenital malformation. It occurs in 0.03–0.7% of births in the United States and most commonly affects Caucasian females. 95% of cases occur without a family history of neural tube defects. After one affected child, the risk increases to 2–5%. It is caused by chromosomal abnormalities or mechanical problems from adhesions to the placenta. The risk is increased with twins, polyhydramnios, hyperthermia, and decreased maternal folate, zinc, or copper. The neonates tend to be stillborn or to die within 2 months. They may have brainstem activity. Anencephaly is associated with the absence of scalp and skull. The brain consists of an exposed mass of tissue with a cerebrovascular area containing vessels, neural tissue, choroid, optic nerves, eyes, some cranial nerves, and brainstem. Craniorachischisis totalis occurs when the entire neuraxis is involved. 15–40% have other organ malformations. Amniotic fluid has increased AFP and acetylcholinesterase. 4. Exencephaly—occurs when the cerebral hemispheres are present but disorganized. There is also absent calvaria and abnormalities of the skull base. 5. Myelomeningocele occurs in 0.07% of births in the United States and is more common in females. It is related to genetic and environmental factors. Risk factors include parental consanguinity, affected siblings, decreased vitamin A or folate, and use of valproic acid or carbamazepine. It is usually in the lumbar spine, with absent arachnoid and adjacent hydromyelia. It is associated with Chiari II malformation (100%), hydrocephalus (80%), lipoma (75%), syringomyelia (50%), diastematomyelia (40%), scoliosis (20%), kyphosis (10%), orthopedic deformities, and callosal dysgenesis (Fig. 3.3). 7. Cephaloceles—include meningocele and meningoencephalocele. In the United States, they are occipital (80%), parietal (10%), frontal (10%), and rarely basal. a. Occipital cephalocele—the most frequent among Caucasians and in Europe and North America. There is a female predilection. The cephalocele protrudes between the foramen magnum and the lambdoid suture. It is associated with myelomeningocele (7%), diastematomyelia (3%), Chiari II and III malformations, Dandy–Walker malformation, and Klippel–Feil syndrome (Fig. 3.4). b. Parietal cephalocele—accounts for 10% of cases in the United States. There is a male predilection. The cephalocele protrudes between the lambda and bregma. It is associated with midline anomalies, agenesis of the corpus callosum, lobar holoprosencephaly, Dandy–Walker malformation, and Chiari II malformations. Fig. 3.2 Spina bifida occulta. Anteroposterior lumbar spine x-ray film demonstrates an L5 bifid spinous process. Fig. 3.3 Myelomeningocele. (A) Neural placode noted on center of dorsal aspect in this neonate prepared for surgery. Note foot deformities and preparation being made for ventriculoperitoneal shunt. (B) Close-up image of the myelomeningocele. Fig. 3.4 Encephaloceles. (A) Two small occipital encephaloceles, and (B) large occipital encephalocele. c. Transsphenoidal cephalocele—rare, associated with sellar abnormalities, endocrine dysfunction, and agenesis of the corpus callosum d. Sincipital (frontoethmoidal) cephalocele—the most frequent type in Southeast Asia and Australian aborigines. There is a male predilection. The cephalocele protrudes between the nasal and ethmoid bones and is not associated with neural tube defects. e. Sphenoethmoidal (nasal) cephalocele—the crista galli is absent or eroded and the foramen cecum is enlarged. The dural diverticulum normally regresses, but if it does not, it may form a dermal sinus tract. They are associated with dermoids, epidermoids, and nasal gliomas (dysplastic or heterotopic glial tissue). If the crista galli is split, there is likely to be a dermoid present (Fig. 3.5). f. Meckel–Gruber syndrome—cystic dysplastic kidneys, cardiac anomalies, orofacial clefting, and cephaloceles. It is associated with maternal hyperthermia on days 20–26 of gestation. 8. Dermal sinus tracts—epithelium-lined tracts caused by faulty segmental disjunction. 60% extend from the skin to the spinal canal, although they may end in the subcutaneous tissue, dura, spinal cord, or nerve roots. 50% end in epidermoids or dermoids. More than 50% are lumbar and the next most frequent site is occipital. There is no gender predilection. Symptoms are mainly a result of infection. They are associated with skin dimples, hyperpigmentation, hairy nevi, and capillary malformations. C. Cleavage disorders (“the face predicts the brain”)—Cleavage of the telencephalon and development of mid-line facial structures are dictated by the prechordal mesoderm. 1. Holoprosencephaly (Fig. 3.6) a. Alobar—completely undivided forebrain, severe craniofacial abnormalities (i.e., cyclopia), absence of olfactory nerves, abnormal optic nerves, monoventricle, fused basal ganglia and thalamus, absence of corpus callosum, septum pellucidum, falx, and fornix. It is associated with trisomy 13, 14, 15, and 18, polydactyly, renal dysplasia, and maternal diabetes. b. Semilobar—monoventricle with forebrain divided by partial falx and interhemispheric fissure, partially separated thalamus and basal ganglia, and variable craniofacial abnormalities (i.e., hypotelorism and cleft lip) Fig. 3.5 Nasofrontal encephalocele. Axial computed tomographic scans and reconstructions (A–D) demonstrate a nasofrontal encephalocele. Fig. 3.6 Holoprosencephaly. Axial computed tomographic scans demonstrate (A) alobar, (B) semilobar, and (C) lobar holoprosencephaly with progressively more cleavage. 2. Arrhinencephaly—the absence of the olfactory bulbs and tracts with normal cortex and gray matter in place of the corpus callosum. It is associated with holoprosencephaly and Kallmann syndrome (anosmia, hypogonadism, and mental retardation). 3. Septooptic dysplasia (de Morsier syndrome)—occurs with mild lobar holoprosencephaly, absence of the septum pellucidum, schizencephaly, and hypoplastic optic nerves. It is associated with seizures, visual symptoms, hypothalamic–pituitary dysfunction (precocious puberty), enlarged ventricles, and hypotelorism (Fig. 3.7). Fig. 3.7 Septooptic dysplasia. Axial computed tomographic scans demonstrate (A) absence of the septum pellucidum and (B, C) thin optic nerves. 4. Cleidocranial dysostosis—occurs with retention of mandibular teeth, delayed closure of fontanelles, wormian bones, and midline defects D. Migrational disorders 1. Normal neuronal migration occurs between the second to fifth gestational months. 2. Heterotopias—normal neurons in abnormal CNS locations (i.e., in the centrum semiovale, along the lateral ventricles or in the cerebellar white matter). They may be laminar or nodular and usually do not enhance (Fig. 3.8). 3. Ectopias—neurons in locations brain tissue should not be in (i.e., the subarachnoid space). They are associated with dysraphism and hydranencephaly. 4. Lissencephaly—smooth brain. In the complete form, the cerebral hemispheres have no sulci. In the incomplete form, there are several shallow sulci. It is associated with in utero infections. 5. Pachygyria—overall decreased number of gyri and those that are present are enlarged 7. Schizencephaly—a gray matter–lined cleft extending from the pia to the ventricle. The cleft may be filled with CSF (opened lip) or collapsed (closed lip) (Fig. 3.9). 8. Porencephaly—a cleft not lined with gray matter but by gliotic white matter that forms after an insult to an otherwise normal brain 9. Unilateral megalencephaly—hamartomatous overgrowth of one hemisphere with ipsilaterally enlarged ventricle and cortex. It is associated with seizures. 10. Agenesis of the corpus callosum—the corpus callosum forms from anterior to posterior, but the rostrum forms last so the splenium and rostrum are almost always involved with agenesis. Agenesis may be complete or partial. It is characterized by a high-riding third ventricle, radial spoke-like gyri, Probst bundles (longitudinal white matter tracts indenting the medial ventricles), and colpocephaly. Occasionally, the cingulate gyrus, anterior commissure, or fornix may be absent. 50% have associ Fig. 3.8 Gray matter heterotopias (arrows). (From Albright AL, Pollack IF, Adelson PD, eds. Principles and Practice of Pediatric Neurosurgery. New York, NY: Thieme; 1999. Reprinted by permission.) Fig. 3.9 Schizencephaly. Sagittal T1-weighted magnetic resonance image demonstrates gray matter-lined cleft extending from the subarachnoid space to the lateral ventricle. Fig. 3.10 Agenesis of the corpus callosum. (A) T1-weighted sagittal and (B) coronal magnetic resonance images demonstrating absence of the corpus callosum with characteristic “Viking helmet” ventricles on coronal view. E. Other developmental syndromes 1. Congenital hydrocephalus—commonly due to aqueductal stenosis, Chiari II malformation, Dandy–Walker malformation, infection, and intraventricular hemorrhage (IVH) 2. Chiari I malformation (Fig. 3.11)—peg-like tonsils extending below the foramen magnum (6 mm below the foramen magnum at age <10 years, 5 mm <30 years, 4 mm <80 years, and 3 mm <90 years). If the tonsils extend >12 mm below the foramen magnum, all are symptomatic and from 5–10 mm, 70% are symptomatic. Chiari I malformation is occasionally acquired after frequent lumbar punctures or placement of a lumboperitoneal shunt. Chiari I malformation is not associated with other brain abnormalities, but is associated with skeletal abnormalities in 25% of cases: basilar invagination (25–50%), Klippel–Feil syndrome with fused cervical vertebrae (5–10%), atlantooccipital fusion (5%), and cervical spina bifida occulta (5%). It presents in early adulthood with pain, occipital headache, Lhermitte sign, long tract signs, syringomyelia (20–40%; with 60–90% of symptomatic cases), and hydrocephalus (25%). Chiari malformations of all types present differently at different age groups. a. Infants—hydrocephalus and brainstem compression with apnea, decreased gag reflex, nystagmus, and spasticity b. Children—nystagmus, spastic paralysis, and bulbar dysfunction c. Adolescents—progressive spasticity and cape-like pain and temperature loss in the upper limbs d. Adults—occipital headache, neck and arm pain, and nystagmus (Fig. 3.11) Fig. 3.11 Chiari I malformation. (A) Sagittal T2- and (B) T1-weighted magnetic resonance image demonstrates peg-like tonsils extending below foramen magnum with associated syringomyelia. 3. Chiari II malformation—thought to develop when the neural folds do not completely meet and there is abnormal ventricular CSF flow into the amnion with collapse of the ventricles. A small posterior fossa develops and as the cerebellum grows, it herniates upward forming a large tentorial incisura and downward pushing the vermis and brainstem through the foramen magnum. It presents in neonates and is associated with many abnormalities (Fig. 3.12). a. Skull and dura—lacunar skull (“lückenschädel,” with scooped out appearance), small posterior fossa, low-lying torcula and transverse sinus, large foramen magnum, concave petrous temporal bones, short concave clivus, and a thin falx cerebri with occasional interdigitating gyri through fenestrations in the falx cerebri (Fig. 3.13) b. Hindbrain—herniation of the vermis, nodulus, uvula, and pyramis through the foramen magnum, medullary kinking (70%), enhancing ectopic choroid, upward cerebellar herniation, and tectal beaking c. CSF spaces—tubular fourth ventricle, large third ventricle with enlarged massa intermedia, colpocephaly (large atria and occipital horns), aqueductal stenosis, small cisterna magna, and hydro-cephalus (90%) Fig. 3.12 Chiari II malformation. Sagittal T1-weighted magnetic resonance image with tectal beaking, low-lying torcula, and vermian descent. Fig. 3.13 Lückenschädel skull. (A, B) Lateral and (C) anteroposterior skull x-ray films demonstrate the scooped out appearance with myelomeningocele. d. Cerebral hemispheres—heterotopias, polymicrogyria, and callosal dysgenesis e. Spine—myelomeningocele (100%), syringomyelia (50–90%), diastematomyelia, and incomplete C1 arch (70%). It is not associated with lipomyelomeningocele. 4. Chiari III malformation—hindbrain herniation into an encephalocele, usually occipital or high cervical (Fig. 3.14) 5. Chiari IV malformation—cerebellar hypoplasia, an entity distinct from the other Chiari malformations 6. Dandy–Walker malformation—posterior fossa cyst continuous with the fourth ventricle with partial or complete vermian absence. It may be due to failure of development of the superior medullary velum (roof of the fourth ventricle) combined with fourth ventricle outlet atresia. It is associated with a large posterior fossa, high tentorium and transverse sinus, lambdoid-torcula inversion, hydrocephalus (80%), callosal agenesis (25%), heterotopias, schizencephaly, cephaloceles, dolichocephaly, cardiac abnormalities, and polydactyly (Fig. 3.15). Fig. 3.14 Chiari III malformation characterized by an occipital encephalocele (curved black arrow) and syringomyelia (thick white arrow). (From Ramsey RG. Teaching Atlas of Spine Imaging. New York, NY: Thieme; 1999. Reprinted by permission.) 7. Dandy–Walker variant—mild inferior vermian hypoplasia with open communication between the fourth ventricle and the cisterna magna through an enlarged vallecula. The fourth ventricle is enlarged, but the posterior fossa is normal size. There may be associated hydrocephalus. Fig. 3.15 Dandy–Walker malformation. (A, B) Sagittal T1-weighted magnetic resonance images and (C) axial computed tomographic scan demonstrate vermian agenesis and connection of the fourth ventricle to a posterior fossa cyst. 8. Mega cisterna magna—posterior fossa CSF accumulation in an enlarged cisterna magna. There is a normal fourth ventricle and no hydrocephalus. It fills with intrathecal contrast. 9. Posterior fossa arachnoid cyst—posterior fossa CSF accumulation with a normal cerebellum and fourth ventricle. There may or may not be hydrocephalus. The cyst does not fill with intrathecal contrast. 10. Lhermitte–Duclos disease—hypertrophied cerebellar granular cell layer and increased myelin in the molecular layer of the cerebellum with thick folia. There is mass effect on the fourth ventricle. The lesion is considered a hamartoma. There may be calcifications, hydrocephalus, and folia with increased signal intensity on T2-weighted MRI. It is associated with Cowden syndrome where patients have facial trichilemmomas, fibromas of the oral mucosa, hamartomatous polyps of the gastrointestinal (GI) tract and breast, and thyroid tumors. Cowden syndrome is due to mutations of the phosphatase and tensin (PTEN) gene on chromosome 10q. 11. Hydromyelia—a distended central canal of the spinal cord lined by ependymal cells. It is associated with Chiari II malformations and myelomeningoceles. 12. Syringomyelia—a fluid-filled cavity in the spinal cord lined by astrocytes 13. Syringobulbia—a fluid-filled cavity in the brainstem 14. Fetal alcohol syndrome—characterized by growth retardation, craniofacial dysmorphism, CNS and visceral malformations, mental retardation, and microcephaly. It is associated with migrational defects, hydrocephalus, schizencephaly, callosal agenesis, and neural tube defects. The incidence is increased if the mother uses drugs, has poor nutrition, or smokes tobacco. 15. Colpocephaly—dilated occipital horns associated with agenesis of the corpus callosum and periventricular leukomalacia, mental retardation, and seizures. 16. Craniosynostosis—premature fusion of the sutures causing abnormal skull growth and shape. Surgical treatment to open the sutures and reshape as needed usually is done between 3–6 months and is generally performed for cosmetic purposes. a. Sagittal synostosis—most common (50%), male predominance, causes scaphocephaly or dolichocephaly (Fig. 3.16) b. Unilateral coronal synostosis—25%, causes anterior plagiocephaly, female predominance (Fig. 3.17) c. Bilateral coronal synostosis—usually genetic, causes brachycephaly, associated with Apert and Crouzon syndromes (Fig. 3.18) Fig. 3.16 Sagittal synostosis (scaphocephaly, dolichocephaly). (A) Computed tomographic (CT) reconstruction and (B) axial CT. Fig. 3.17 Unilateral left coronal synostosis. Computed tomographic reconstruction with “harlequin eye” on the left. Fig. 3.18 Brachycephaly due to bilateral coronal synostosis. d. Unilateral lambdoid synostosis—1.3%, male predominance, causes posterior plagiocephaly and must be distinguished from the much more common positional plagiocephaly resulting from a baby lying on one side too often. Differentiating the two conditions can often be accomplished by history and physical examination (true synostosis is present at birth, positional is not). In positional plagiocephaly, a parallelogram-type deformity is appreciated when the head is viewed from above and associated with the following: unilateral flattening of the occiput, ipsilateral frontal/parietal bossing, ipsilateral cheekbone prominence, ipsilateral anterior displacement of the ear, and contralateral occipital bossing. This is treated initially by changing the infant’s sleeping position, not by surgery. Unilateral lambdoid synostosis is also characterized by unilateral flattening of the occiput, ipsilateral frontal/parietal bossing (typically less severe), and contralateral occipital bossing. In contradistinction, the ipsilateral ear is typically posteriorly and inferiorly displaced and the posterior skull base is tilted with an unusually prominent ipsilateral mastoid process (Fig. 3.19). e. Metopic synostosis—5%, causes trigonocephaly (Fig. 3.20) f. Turricephaly—5–10%, synostosis of both coronal sutures and the sphenofrontal sutures, causes a tall broad head g. Oxycephaly—5–10%, synostosis of multiple sutures, causes a cone-shaped head h. Pansynostosis—causes Lückenschädel head i. Crouzon syndrome—most frequent craniofacial syndrome, autosomal dominant or sporadic inheritance, shallow orbits, exophthalmos, midface hypoplasia, malformed ears, agenesis of the corpus callosum, less severe mental retardation than with Apert syndrome, and increased incidence of hydrocephalus. More than one suture is involved, and they may have oxycephaly, turricephaly, or dolichocephaly. The sphenofrontal synostosis produces exophthalmos; associated with mutations of the fibroblast growth factor receptor 2. j. Apert syndrome—second most common craniofacial syndrome, autosomal dominant or sporadic inheritance, turricephalic head, maxillary hypoplasia, orbital hypertelorism, syndactyly, mental retardation, deafness, flat nose, and vertebral and skeletal abnormalities. Only the coronal suture is involved. There are GI, GU, and cardiac abnormalities, and increased incidence of frontal encephaloceles; also associated with mutations of the fibroblast growth factor receptor. Fig. 3.19 Lambdoid synostosis as demonstrated on axial computed tomographic scan. Fig. 3.20 Metopic synostosis (trigonocephaly); (A) axial computed tomographic (CT) scan and (B) CT reconstruction. 17. Klippel–Feil syndrome—congenital fusion of the upper cervical vertebrae; associated with Sprengel deformity (elevation of the scapula) and Chiari I malformation (Fig. 3.21) F. Chromosomal disorders 1. Trisomy 13 (Patau syndrome)—female predominance, hypotelorism, holoprosencephaly, microcephaly, microphthalmia, cleft palate and lip, polydactyly, dextrocardia, and ocular abnormalities. Death ensues before 9 months. 2. Trisomy 18 (Edward syndrome)—female predominance, gyral dysplasia, callosal agenesis, Chiari II malformation, dolichocephaly, cerebellar hypoplasia, hypertelorism, microphthalmia, syndactyly, rocker bottom feet, and ventricular septal defects. Less than 10% live 1 year. 3. Trisomy 21 (Down syndrome)—most frequent of the chromosomal abnormalities and occurs in 0.1% of births. Varieties include (1) sporadic trisomy (95%) (increased risk with maternal age >35 years), (2) Robertson translocation 21q and 14 or 21q and 22q in 4% (not associated with age), and (3) mosaic (1%). It is characterized by brachycephaly, hypotelorism, hypoplastic maxilla and nose, simian crease, epicanthal folds, skull base abnormalities, cervical stenosis, atlantoaxial instability, underdeveloped inferior temporal gyrus, narrow superior temporal gyrus, flattened occipital pole, mental retardation, lens opacities, congenital heart disease (increased risk of brain abscess), and Alzheimer disease (by 40 years). Recall: Amyloid precursor protein is found on chromosome 21. 4. 5p deletion (cri-du-chat syndrome)—mental retardation, micro-cephaly, hypertelorism, and congenital heart disease 5. 15q deletion (Prader–Willi syndrome)—mental retardation, truncal obesity, short stature, and hypogonadism 6. Fragile X syndrome—the most frequent hereditary cause of mental retardation. It is more common (and severe) in males (1 in 4,000) than females (1 in 8,000). There is a dysmorphic appearance with long face, large testicles, and vermian hypoplasia. The syndrome is caused by mutation of a single gene, the FMR1 (Fragile X Mental Retardation 1) gene. This mutation is characterized by massive expansion of a trinucleotide repeat that leads to transcriptional shutdown of the gene. Fig. 3.21 X-ray showing KlippelFeil syndrome. A. Perinatal brain injury—may be caused by birth or other trauma, infection, metabolic disorders, or maternal intoxications B. Caput succedaneum—cutaneous hemorrhagic edema in the skin over the calvarium caused by pressure during birth with vascular stasis. It crosses sutures and resolves in 48 hours. C. Subaponeurotic hemorrhage—Blood accumulates under the aponeurosis. D. Cephalohematoma—subperiosteal blood that does not cross suture lines. It is usually parietal and may calcify. E. Epidural hematoma (EDH)—rare, associated with fractures F. Subdural hematoma (SDH)—rare, caused by mechanical trauma from a small birth canal, rapid or prolonged labor, abnormal presentation, premature birth with increased skull compliance, large head, and forceps deliveries. It forms from tearing of the bridging veins over the convexity, tentorium, or skull base (Fig. 3.22). Occipital osteodiastasis is the traumatic separation of the squamous and lateral occipital bone with tearing of the occipital sinus and bleeding in the posterior fossa. Laceration of the falx or inferior sagittal sinus causes bleeding over the corpus callosum. There is a subgroup of falcine or tentorial SDH associated with asphyxia or mechanical trauma that are usually small and rarely have clinical significance. G. Subarachnoid hemorrhage (SAH)—caused by hypoxia or trauma and usually has a good outcome H. Intraparenchymal hemorrhage—caused by coagulation defects, vitamin K deficiency, trauma, vascular malformation, tumor, or stroke Fig. 3.22 Subdural hematoma (SDH). Nonenhanced computed tomographic scans demonstrate (A) bilateral subacute SDH, (B) chronic left SDH with acute hemorrhage (fluid-fluid level), and (C) posterior fossa SDH. J. Delivery trauma—usually causes SDH (interhemispheric or tentorial) and less frequently posterior fossa SAH that results from the sudden frontooccipital shortening that may tear a sinus or vein. The risk increases with cephalopelvic disproportion or abnormal presentation. K. Hypoxic/ischemic perinatal brain injury 1. Acute injury a. Periventricular leukomalacia—increased incidence with prematurity, congenital heart disease, shock, sepsis, and respiratory distress syndrome. It occurs within 72 hours and causes lower extremity weakness and visual problems. b. Gray matter necrosis—seen in full-term births with perinatal asphyxia and mainly affects the subthalamus, lateral geniculate body, inferior colliculus, cranial nerves, dentate nuclei, Purkinje cells, and the cortical internal granular layer 2. Chronic injury a. Ulegyria—parietooccipital mushroom-like atrophic gyri with atrophy in sulcus valleys b. Status marmoratus—the thalamus, neostriatum, and cortex develop irregular intersecting bands of myelin and astrocytic fibers that grossly resemble marble caused by cell loss followed by remyelination 3. Kernicterus—caused by increased unbound unconjugated bilirubin staining the gray matter and causing neuronal necrosis. It affects predominantly the GP, thalamus, subthalamus, and CNs III and VIII nuclei, causing symmetric cell loss with extrapyramidal motor signs. Treat the elevated bilirubin with phototherapy using ultraviolet light, but if the bilirubin is >20 with associated sepsis, prematurity, acidosis or low albumin, treat with an exchange transfusion. 4. Cavitary encephalopathies—caused by infections, genetic defects, or toxic gases a. Porencephaly—a cavity that extends from the leptomeninges to the ventricles or superficial white matter lined by white matter (as opposed to schizencephaly, which is a cleft lined by gray matter). b. Hydranencephaly—most of the cortex is replaced by CSF, may result from cerebral ischemia (internal carotid artery [ICA] occlusions) or infection (e.g., CMV or toxoplasmosis) (Fig. 3.23). c. Multicystic encephalopathy—ACA and middle cerebral artery (MCA) distribution, multiple cavities A. Meningitis 1. Etiologies—the most frequent pathogen overall is Haemophilus influenzae, and the most frequent cause in adults is Streptococcus pneumoniae. The subarachnoid space near the blood vessels fills first with neutrophils and fibrin, then macrophages, and then it eventually fibroses. Meningitis is associated with arteritis, phlebitis, superior sagittal sinus thrombosis, and hydrocephalus. The three most common causes of meningitis (H. influenzae, S. pneumoniae, and Neisseria meningitidis) all normally colonize the nasopharynx. The fourth most common pathogen is Listeria, which causes opportunistic infections. Staphylococcus aureus is associated with postoperative infections, and S. epidermidis is the most common cause of shunt infections. The mortality from various types of meningitis is 10% (H. influenzae and N. meningitidis), 25% (S. pneumoniae), and 50% (neonatal, with 50% of survivors having permanent sequelae). Fig. 3.23 Hydranencephaly; axial computed tomographic scan demonstrates a very thin rim of brain. 2. Neonate (0–4 weeks)—the most frequent organisms are (1) group B streptococcus with acute onset <5 days post-partum with sepsis and pneumonia. It is associated with intrapartum infection and has a 15–30% mortality; (2) Escherichia coli is the most frequent cause in Latin America, especially with the K1 capsular antigen; and (3) Listeria. Sequelae include hydrocephalus, encephalomalacia at the border zones, and in the white matter, seizures, deafness, mental retardation, and focal deficits. The overall morbidity and mortality is 30–50%. Risk increases with prematurity, prolonged membrane rupture, traumatic delivery, congenital malformations (myelomeningocele or dermal sinus), and acquired respiratory, GI, and umbilical infections. 3. 4 to 12 weeks—The most common pathogen is S. pneumoniae. 4. 3 months to 3 years: The most common pathogen is H. influenzae, although it rarely occurs after 5 years; 85% are type b. It develops from nasopharyngeal colonization that leads to sepsis followed by meningitis. Blood cultures are positive in 70%. The bacteria need X and V growth factors to grow. It is associated with bilateral subdural effusions (usually culture negative because they are due to increased permeability of blood vessels, also seen with S. pneumoniae infections) and frequently causes seizures. Treatment is with third-generation cephalosporins. Concomitant use of steroids has been shown to decrease the incidence of deafness in children. 5. Children and young adults—the most frequent pathogen is N. meningitidis, and the meningitis occurs in epidemics. It develops from nasopharyngeal colonization that leads to hematologic dissemination and meningitis. The risk increases with decreased complement and in patients with systemic lupus erythematosus (SLE). It is associated with cutaneous eruptions (60%), arteritis, and cardiac deaths. The Waterhouse–Friderichsen syndrome is meningococcemia with vasomotor collapse, shock, diffuse intravascular coagulation, and diffuse hemorrhages, especially in the adrenal glands. N. meningitidis is a gram-negative intracellular diplococcus. Treatment is with penicillin or chloramphenicol. 6. Elderly—the most common pathogen is S. pneumoniae. It is more common in the elderly (because of age-dependent immune decline) and alcoholics (ethanol induces a chemotaxis defect and impairs phagocytosis). Risk is increased with trauma (it is the normal flora of the mastoid, ear, sinus, and nose), infection and sickle cell disease (caused by impaired splenic filtering, the risk is not increased in adults). Detection uses the Quellung reaction. Treatment is with penicillin. B. Brain abscess (Fig. 3.24 and Fig. 3.25) 1. Etiologies—the most common pathogen is Streptococcus and the most common cause is adjacent ear or sinus infection (40%), with spreading along the valveless venous channels (frontal sinus to frontal lobe, sphenoid sinus to temporal lobe, and ear to cerebellum or temporal lobe). 33% are from hematogenous spread from another infected site (usually lung, dental disease, or trauma, more likely multiple; and usually in the distal MCA territory at the gray/white matter junction); 20% have no identifiable cause.spread from another infected site (usually lung, dental disease, or trauma, more likely multiple; and usually in the distal MCA territory at the gray/white matter junction); 20% have no identifiable cause.spread from another infected site (usually lung, dental disease, or trauma, more likely multiple; and usually in the distal MCA territory at the gray/white matter junction); 20% have no identifiable cause.spread from another infected site (usually lung, dental disease, or trauma, more likely multiple; and usually in the distal MCA territory at the gray/white matter junction); 20% have no identifiable cause. Fig. 3.24 Brain abscess. (A) Nonenhanced and (B) enhanced axial computed tomographic scans demonstrate a smooth ring-enhancing lesion. (C) T2-weighted magnetic resonance image demonstrates the characteristic hypointense capsule and surrounding hyperintensity, which is the associated edema. (D,E) There is a thin enhancing rim on T1-weighted MRI with gadolinium and (F) restricted diffusion on the diffusion-weighted image. Fig. 3.25 Neonatal brain abscess. (A) Axial T1-weighted nonenhanced and (B) enhanced magnetic resonance images (MRIs) and (C) enhanced sagittal T1-weighted MRI demonstrate a massive cystic abscess cavity. 3. Neonates—The most common causes are Citrobacter, Bacteroides, Proteus, and gram-negative bacilli. (Fig. 3.25). 4. Trauma—The most frequent cause is Staphylococcus, and with otitis, enteric bacilli are common. 5. Subacute bacterial endocarditis (SBE)—with streptococcal deposits on the valves usually causes ischemic stroke, but not abscesses. 10% of SBE cases develop infectious aneurysms. Acute endocarditis caused by Staphylococcus or hemolytic Streptococcus may be associated with multiple abscesses. There are mixed species in 30%. 6. Brain abscesses occur most commonly at the gray–white matter junction and are multiple in 35%. The abscess has a tendency to rupture into the ventricle because the capsule is thinner medially where there is decreased collagen formation. This has been postulated to be due to increased oxygen content near the cortex that stimulates the fibroblasts to develop. The mortality is 5% and is mainly from brain herniation or abscess rupture into the ventricles. 7. Pathologic stages— (a) early cerebritis (up to 5 days), (b) late cerebritis (5 days–2 weeks); (c) early capsule formation (2–3 weeks); and (d) late capsule formation (>3 weeks, there is a firm capsule around the abscess). C. Encephalitis—commonly by Legionella (CSF culture is usually negative), Mycoplasma, Listeria (in neonates and immunosuppressed, CSF culture is usually positive), and Brucella D. Subdural empyema—mainly caused by Streptococcus and Bacteroides from an adjacent infection. Treat with antibiotics and drainage with multiple burr holes or osteoplastic flap and consider antibiotic therapy alone if asymptomatic (Fig. 3.26). E. Epidural abscess—most commonly caused by S. aureus, Streptococcus, gram-negative bacilli, and tuberculosis (TB). It is usually secondary to an associated osteomyelitis or sinusitis and most frequently occurs in the thoracic, lumbar, or sacral spine (Fig. 3.27). F. Osteomyelitis—most commonly caused by the same bacterial species as epidural abscesses and occurs with sinusitis (Fig. 3.28), after craniotomy or by hematologic spread. It is characterized by moth-eaten cortical bone with poor margins and soft tissue swelling. X-ray films may appear similar to metastatic lesions. Gradenigo syndrome is petrous apex osteomyelitis with CN VI palsy and retroorbital pain and may occur in children from extension of severe otitis. Fig. 3.26 Subdural empyema; thick pus collection over the convexity. Fig. 3.27 Epidural abscess and subdural empyema. Enhanced computed tomographic scan demonstrates a large convexity, low-density subdural collection and a small anterior rim-enhancing epidural lesion (arrow). Fig. 3.28 Sphenoid sinusitis. (A) Axial nonenhanced and (B) enhanced T1-weighted magnetic resonance images demonstrating thickened enhancing mucosa. A. Mycobacterial infections 1. TB—caused by Mycobacterium tuberculosis a. CSF—increased lymphocytes, decreased glucose (but not as low as with pyogenic infections) and increased protein up to 200 mg/dL. Acid-fast bacilli are seen in the CSF in <25% of cases and require up to 4 weeks to grow in culture. It is diagnosed by positive purified protein derivative (PPD) skin test and TB lesions found elsewhere in the body. b. TB meningitis—characterized by a thick basilar exudate, small miliary granulomas on the convexities, and frequent vascular occlusions. Morbidity is 80% and mortality is 30%. It usually results from hematogenous spread. Pathologic examination (Fig. 3.29) demonstrates granulomas with caseating necrosis, lymphocytes, and Langerhans giant cells. c. Tuberculomas—In the brain, parenchyma are rare in the United States, but are common intracranial masses in India and Mexico. They may be solid or cystic, 30% are multiple; in children, two thirds are infratentorial. d. Miliary TB—has innumerable small lesions, is rare, and is more frequently seen in children e. Two-thirds of cases have active TB infections elsewhere in the body. Treatment is with 24 months of triple antibiotics. 2. Leprosy—caused by Mycobacterium leprae. It most frequently occurs in tropical climates, California, Texas, New York, and Louisiana. In the United States, it is primarily brought in by travelers. There are two types: Fig. 3.29 Tuberculous meningitis (hematoxylin and eosin [H&E] stain); caseating granuloma with Langerhans giant cells. a. Lepromatous form seen with low host resistance and has lepra cells that are plump histiocytes filled with organisms. Lesions are located in the skin on cooler parts of the body (i.e., hands, feet, head, peripheral nerves, anterior eyes, upper airways, and testes). The lepromin skin test is negative. b. Tuberculoid form is seen with maximal host resistance, and there are areas of hypesthetic skin, inflamed swollen nerves, caseating granulomas, and occasional gram-negative bacilli seen on histologic examination. The lepromin skin test is positive. B. Other granulomatous disease 1. Sarcoid is a systemic granulomatous disease of unknown cause that involves lymph nodes (especially hilar), lungs, skin, eyes, salivary glands, and liver. The nervous system is involved in 5% of cases with cranial nerve palsies (especially CN VII), aseptic meningitis, pituitary dysfunction, hydrocephalus, and noncaseating granulomas at the base of the brain (especially the hypothalamus). It may also cause myelopathy, neuropathy, and myopathy. Sarcoid is most common in African Americans. The serum angiotensin converting enzyme (ACE) level usually is elevated. Treatment is with steroids (Fig. 3.30). 2. Whipple disease—a chronic multisystem disease caused by Tropheryma whippelii. It is characterized by weight loss, abdominal pain, diarrhea, lymphadenopathy, arthralgia, and Alzheimer disease–like neurologic symptoms (10%). Pathologic examination demonstrates foamy macrophages with PAS-positive granules. These are degenerating bacilli. Treatment is with tetracycline. C. Spirochetes 1. Neurosyphilis—a sexually transmitted disease caused by Treponema pallidum. A genital lesion (chancre) forms 3 weeks after infection and secondary lesions develop several weeks later. 25% of cases involve the CNS, but usually after 3 years. Treatment is with penicillin and CSF testing at 6 months and 1 year for evaluation of treatment success. There are four types of neurosyphilis: a. Meningovascular syphilis (lues)—occurs after 7 years and is characterized by subacute or chronic meningitis with perivascular lymphocytes, Huebner arteritis with intimal proliferation and vessel obliteration, and multiple ischemic strokes in the basal ganglia and MCA territory. Fig. 3.30 Neurosarcoid. (A) Nonenhanced and (B) enhanced sagittal T1-weighted magnetic resonance imaging scans demonstrate the sellar and suprasellar enhancement. b. General paresis of the insane—occurs after 15 years and is characterized by invasion into the parenchyma with chronic encephalitis, atrophy, and gummas with nonsuppurative necrotic debris, progressive physical and mental deterioration, and Argyll Robertson pupils. c. Tabes dorsalis—occurs after 15–20 years and is characterized by myelopathy from meningeal fibrosis. There is mainly dorsal root and posterior column involvement, W-shaped demyelination in the thoracic and lumbar spinal cord from the posterior horns inward, lightning-like pains, sensory ataxia, urinary incontinence, decreased lower limb deep tendon reflexes, decreased proprioception and vibratory sense, positive Romberg test, Argyll Robertson pupils (90%), ptosis, optic atrophy, and Charcot joints of the hip, knee, and ankle. d. Congenital syphilis—includes Hutchinson triad of notched teeth, deafness, and interstitial keratitis. This is also associated with meningovascular syphilis. 2. Lyme disease—caused by Borrelia burgdorferi, a tick-borne spirochete that causes erythema migrans (70%). 15% have neurologic symptoms such as aseptic meningitis, cranial neuritis (especially CN VII), encephalitis, myelopathy, radiculopathy, and peripheral neuropathy. It is most frequently seen from May through July in New England, along the Pacific coast, and in Wisconsin and Minnesota. Symptoms are caused by immune complexes and vasculitis with a postviral-type demyelination and perivascular infiltration. Diagnosis is by enzyme-linked immunosorbent assay (ELISA) and treatment is with tetracycline. D. Other bacterial infections 1. Actinomyces—a branched filamentous bacterium that looks like a fungus. It is rare in the CNS, contains sulfur granules, and is acid fast bacillus (AFB) negative. 2. Nocardia—an opportunist infection that usually infects the lungs, although the CNS is involved in 30% of cases. It forms multiple abscesses and is AFB positive. E. Fungal infections 1. Mycotic infections—usually occur in immunosuppressed patients and result from hematologic spread from the lungs. They cause basilar meningitis (with cranial neuropathies, hydrocephalus, and arteritis causing strokes) or abscesses with granulomas. CSF reveals increased lymphocytes and decreased glucose. All fungi stain with methenamine silver. The true pathogens are Histoplasma, Blastomyces, and Coccidiomycoses; all others only infect the immunosuppressed. 3. Histoplasmosis—caused by H. capsulatum from the soil of the Ohio, Mississippi, and St. Lawrence River valleys. It frequently causes pulmonary infections and only rarely CNS infections with basilar meningitis and occasional parenchymal granulomas. 4. Blastomycosis—caused by B. dermatidis from the soil of the eastern United States. The CNS is involved in less than 5% of cases. Abscess is more frequent than meningitis. Culture demonstrates a single, budding yeast. 5. Cryptococcosis—caused by C. neoformans from soil contaminated with bird feces. It is the most frequent fungal meningitis and the second most frequent CNS fungal infection. There are rarely cryptococcomas in the parenchyma. CSF cultures are positive (75%), Cryptococcus antigen is positive (90%), and India ink stain is positive (Fig. 3.31) (50%), with a single budding yeast with a thick capsule. The capsule disappears during the tissue preparation to form the characteristic halo. Treatment is with amphotericin B and fluconazole. Fig. 3.31 Cryptococcus (India ink stain); yeast organism surrounded by clear halos. 6. Coccidioidomycosis—caused by C. immitis from the soil of the Southwest United States, especially in California (the San Joaquin Valley) and Arizona. The CNS is involved in 63% of cases with meningitis or cerebritis with granulomas. There are large sporangia filled with endospores. Treatment is with intrathecal amphotericin B and mortality is 50%. 7. Aspergillosis—caused by Aspergillus fumigatus or A. flavus from the soil. It is the third most frequent fungal CNS infection. It is named from its sporangia that resemble a container of holy water and only forms when growing with contact to air. It is rarely diagnosed before death. It is angioinvasive causing hemorrhagic cerebritis and rarely meningitis. It has branching septate hyphae. Treatment is with amphotericin B and 5-fluorocytosine (Fig. 3.32 and Fig. 3.33). F. Parasites 1. Nematodes (roundworms) Fig. 3.32 Aspergillus encephalitis (hematoxylin and eosin [H&E] stain); hemorrhagic cerebritis with branching septate hyphae. Fig. 3.33 Aspergillus (silver stain); branching septate hyphae. b. Angiostrongylus cantonensis—a rat lung worm that involves the CNS causing eosinophilic meningitis c. Strongyloidiasis—caused by the roundworm, Strongyloides stercoralis. This worm can carry bacteria with it to the CNS and cause mixed meningitis 2. Platyhelminth (flatworms) a. Cestodes (tapeworms) (1) Cysticercosis—caused by Taenia solium, the pork tapeworm. It is the most frequent CNS parasitic disease in the world. The human is the definitive host of the adult worm. Cysticercosis occurs when a human serves as the intermediate host and the cysticerci accumulate in the subcutaneous tissue, skeletal muscles, eyes, and CNS. It may cause meningitis, seizures, parenchymal or intraventricular abscesses. Taeniasis (tapeworm infection) is contracted by ingestion of undercooked pork, whereas cysticercosis is contracted by ingestion of food with fecal contamination. Treatment is with praziquantel or albendazole (Fig. 3.34–Fig. 3.36). (2) Echinococcus (hydatid disease)—caused by E. granulosa, a dog tapeworm. The intermediate host is sheep. Each cyst contains multiple larvae (hydatid sand), and rupture can lead to further cyst formation. Cysts form in the liver (65%), lung (20%), and brain (2%). Treatment is with mebendazole. b. Trematodes (flukes) (1) Schistosomiasis—caused by S. haematobium and S. mansoni, which can infect the spinal cord and by S. japonicum, which can infect the brain. They live in blood vessels and the snail is the intermediate host. Treatment is with praziquantel. (2) Paragonimiasis—caused by a fluke that resides in the lung and rarely enters the brain (1%) by way of the basal foramina. It can be acquired by ingestion of raw fish. G. Protozoa 1. Toxoplasmosis—caused by T. gondii, an obligate intracellular organism. The definitive host is the cat. Human infection is caused by ingestion of cat feces or raw meat. Serum antibodies are seen in 30% of the normal population. The cyst may lie dormant until the host becomes immunocompromised. It is the most frequent mass lesion in the brain in patients with AIDS. It is hypoactive on thallium-201 single photon emission CT. The cyst is full of and is surrounded by free tachyzoites that can be seen on H & E. Infection of the mother during pregnancy causes congenital lesions such as brain necrosis, periventricular calcification, hydrocephalus, hydranencephaly, chorioretinitis, and hepatosplenomegaly. Acquired infection may cause meningoencephalitis or abscesses. Treatment is with 4 weeks of sulfadiazine and pyrimethamine with leucovorin (to rescue the cells from the antifolate effects) or for the remainder of life in AIDS patients (Fig. 3.37). Fig. 3.34 Cysticercosis. Non-enhanced sagittal T1-weighted magnetic resonance images demonstrate cystic lesions with a scolex (A) in the left temporal lobe and (B) in the fourth ventricle. Fig. 3.35 Cysticercosis (gross); cyst with larva. Fig. 3.36 Cysticercosis (hematoxylin and eosin [H&E] stain); encysted larva with scolex. 2. Amoebic meningoencephalitis—caused by (1) Naegleria fowler from freshwater ponds or lakes that enters the skull through the cribriform plate and causes basilar hemorrhagic meningitis that may be rapidly fatal, (2) Acanthamoeba from contact lenses that gets in the corneal stroma (treatment may require a corneal transplant), and (3) Entamoeba histolytica that also may cause meningoencephalitis. Fig. 3.37 Toxoplasma. Pseudocyst and multiple free tachyzoites in the brain (hematoxylin and eosin [H&E] stain). 3. Malaria—caused mainly by Plasmodium falciparum, but also by Plasmodium vivax, Plasmodium malariae, and Plasmodium ovale. One to 10% of cases involve the CNS and manifest as acute encephalopathy, fever, decreased mental status, seizures, and focal deficits. Symptoms may be due to cerebral hypoxia from capillary obstruction by infected erythrocytes. 4. Trypanosomiasis—caused by (1) Trypanosoma brucei that causes African sleeping sickness, a meningoencephalitis transmitted by the tsetse fly; (2) T. rhodesiense that causes a more severe meningoencephalitis; (3) T. gambiense that is more chronic; and (4) T. cruzi that causes Chagas disease in South America, which is transmitted by the reduviid bug and causes cardiomyopathy, megacolon, and congenital CNS lesions in children. H. Viruses 1. Viruses—obligate intracellular parasites that depend on the host cell for protein synthesis and energy. They may contain either DNA or ribonucleic acid (RNA). They have no organelles or nucleus and are not cells. They are surrounded by a capsid with protein subunits (capsomeres). 2. Neurotropic RNA viruses—picornavirus (poliomyelitis), togavirus (Eastern and Western equine encephalopathy and rubella), flavivirus (St. Louis encephalopathy), paramyxovirus (measles, SSPE), rhabdovirus (rabies), arena (lymphocytic choriomeningitis), bunyavirus (California group encephalitis), and retrovirus (human immunodeficiency viruses [HIV]) 3. Neurotropic DNA viruses—papovavirus (PML) and the herpes group with HSV-1 and 2, varicella-zoster, and CMV 4. Infection transmission a. Respiratory route—measles, mumps, and varicella-zoster b. GI route—poliovirus c. Subcutaneous inoculation—rabies and arbovirus 5. Virus access to the CNS a. Blood—poliovirus b. Peripheral nerves—rabies a. Circular without envelope—papovavirus JC and SV40 b. Circular with envelope—poxvirus c. Linear without envelope—adenovirus d. Linear with envelope—herpesvirus 7. RNA viruses a. Single strand, with sense, without envelope—picornavirus (poliovirus, echovirus, and Coxsackie virus) b. Single strand, with sense, with envelope—togavirus (rubella and eastern, western, and Venezuelan equine encephalopathies), retrovirus (HIV), and flavivirus (St. Louis and Japanese encephalopathies) c. Single-strand, without sense, with envelope—paramyxovirus (measles, SSPE, mumps), rhabdo-virus (rabies), bunyavirus (California encephalopathy), orthomyxovirus (influenza), and arenavirus (lymphocytic choriomeningitis) 8. Virus detection—by in situ hybridization with radioactive complementary DNA or RNA or by polymerase chain reaction (PCR) used to amplify their DNA segments. HSV is persistent so it can always be recovered. Varicella-zoster becomes latent so it is intermittently recovered. 9. Viral (aseptic) meningitis—70% are caused by enterovirus (picornavirus: polio, echovirus, Coxsackie virus) and mumps. It is self-limited and usually lasts up to 1 week. a. Symptoms—fever, headache, and nuchal rigidity. There are no features of encephalitis such as decreased mental status, seizures, or focal deficits. b. CSF evaluation—lymphocytic pleocytosis, normal protein, and glucose (except decreased glucose with mumps and lymphocytic choriomeningitis virus in rodent handlers) and no bacteria or fungus on culture. c. Epidemiology—it is most common in August and September. Nonviral aseptic meningitis may be caused by an adjacent sinus or mastoid bacterial infection, syphilis, Cryptococcus, TB, Borrelia, leukemia, lymphoma, and Behçet disease. 10. Viral encephalitis—the most frequent epidemic cause is arbovirus and the most frequent sporadic cause is HSV-1. In immunocompromised patients, the most common viruses are HIV, CMV, and papovavirus (there are also infections from Toxoplasma, Aspergillus, and Listeria). Symptoms include seizures, decreased mental status, and focal deficits. There is a 5–20% mortality (HSV has 50% mortality) and 20% incidence of permanent sequelae. 11. Arboviruses—“Arthropod borne” by mosquitoes, ticks, etc. Reservoirs are birds, small mammals, and horses. Humans are dead-end hosts. They include togavirus, flavivirus, bunyavirus, and reovirus. a. Eastern encephalopathy—the most severe and occurs in late summer on the eastern seaboard of the United States and has a 70% mortality b. Western encephalopathy—milder and occurs on the west coast of Canada, the United States, and Central America c. Venezuelan equine encephalopathy—mild, occurs in Central and South America and has <1% mortality d. LaCrosse encephalitis—the second most frequent encephalitis in the United States after entero-virus, has a low mortality, and occurs in the Midwest and New York e. St. Louis encephalitis—transmitted by birds and occurs in the summer in the Midwest and South g. Pathologic findings of all these viruses—perivascular mononuclear infiltrates 12. Herpes encephalitis a. HSV—enters through the eye, mouth, and genitals and reaches the CNS by the peripheral nerves. In the CNS, it remains relatively shielded from the host’s immune system. (1) The virus may travel from the skin to the olfactory nerve, trigeminal nerve, or trigeminal ganglion. It rarely spreads hematogenously. It is found in the trigeminal ganglion in 50% of normal adults and reactivates after trauma or immunosuppression. (2) Findings—aseptic meningitis, encephalitis, myelitis, keratitis, skin lesions, and hemorrhagic necrosis of the basal frontal and medial temporal lobe s (Fig. 3.38) (3) It is generally held that in children, the virus reaches the CNS by means of the olfactory mucosa and in adults by means of the trigeminal ganglion to the middle fossa. HSV-1 is the most frequent cause of sporadic encephalitis. (4) It usually affects those older than neonates, frequently involves limbic structures, and sequentially becomes bilateral. HSV-2 encephalitis affects mainly neonates and is more diffuse. Fig. 3.38 Herpes encephalitis. Axial magnetic resonance images (MRIs) demonstrate hemorrhagic and edematous left medial temporal lobe lesion. (A) Nonenhanced T1-weighted MRI, (B) enhanced T1-weighted, (C) proton density, and (D) T2-weighted MRIs. (6) Pathologic examination (Fig. 3.39)—demonstrates Cowdry type A inclusions (intranuclear eosinophilic masses with a surrounding halo found in neurons, oligodendrocytes, and astrocytes). These inclusions are best seen in the first few days of the infection. The CSF culture is usually negative. Mortality is 50–60%. Treatment is with steroids and acyclovir IV for 14 days. Fig. 3.39 Herpes simplex virus-1 encephalitis (hematoxylin and eosin [H&E] stain); lymphocytic perivascular cuffing. Inset with Cowdry A eosinophilic intra-nuclear inclusions. b. CMV—the largest herpesvirus. It is commonly found in all body fluids. It also produces Cowdry type A and intracytoplasmic inclusions. Diagnosis is by culture and in situ hybridization. Intrauterine infections cause congenital malformations such as microcephaly, hydrocephalus, chorioretinitis, and microphthalmia (Fig. 3.40). Fig. 3.40 Cytomegalovirus (hematoxylin and eosin [H&E] stain); eosinophilic intra-nuclear and intracytoplasmic inclusions. (1) Herpes infection of the geniculate ganglion (Ramsay Hunt syndrome)—may cause dysfunction of CN VII with altered sensation of taste, facial weakness, and vesicular eruptions on the pinna and in the external auditory canal (2) Pathologic examination—intranuclear inclusions in the DRG and the posterior horn gray matter, posterior roots, and meninges (3) Treatment—skin lotions (calamine), capsaicin, acyclovir (must be given within 48 hours of rash formation to decrease the duration of disease, use 7-day oral course or 10-day IV course for immunocompromised patients or if >3 dermatomes involved) and topical acyclovir for the eye with a V1 infection. Postherpetic pain is treated (although it responds poorly) with amitriptyline, carbamazepine, gabapentin, pregabalin, and time. 13. Rabies—acquired from the bites of skunks, foxes, coyotes, and bats. It may also be transmitted by aerosolized vectors inside caves where bats live. The virus travels through the peripheral nerves to the CNS and has an incubation period of 1 to 3 months. a. The prodrome phase is followed by either a paralytic (Guillain–Barré-like) form or encephalitic (more frequent) form that progresses to coma and death 2 to 25 days after the onset of symptoms. b. The virus destroys mainly limbic neurons. Negri bodies (intracytoplasmic eosinophilic collections of ribonucleoproteins) are seen in 80% of cases and are especially prominent in the cerebellum (Purkinje cells), brainstem, and hippocampus (Fig. 3.41). c. Symptoms—anxiety, dysphagia, and spasms of the throat when attempting to swallow (hydro-phobia) d. Treatment—washing the wound with soap, water, and benzyl ammonium chloride (inactivates virus) and watching the biting animal for 10 days. If it becomes symptomatic, it should be euthanized and the brain examined. If there are signs of rabies, treat with rabies immune globulin to provide 10 to 20 days of passive immunization. If one is at high risk, consider a vaccine. Fig. 3.41 Rabies (hematoxylin and eosin [H&E] stain. Large intracytoplasmic eosinophilic Negri body in Purkinje cell between the granular and molecular layers. a. Poliovirus (single-stranded RNA virus) (1) There are <10 cases/year in the United States and they are mostly vaccine related. Incubation is 7 to 21 days. 10% of cases develop viremia. (2) Most infections are subclinical (nonparalytic form). The CNS is involved in 0.1–1% of cases with aseptic meningitis, encephalitis, and paralysis. (3) Anterior horn cells—the most susceptible because they have increased numbers of viral receptors on their surface. The virus may also infect Betz cells and brainstem nuclei (10–35% of patients develop bulbar palsy). (4) Pathologic examination reveals perivascular mononuclear infiltrates and neuronophagia. There is no virus detected in the CSF. (5) The risk of paralytic disease with the live attenuated oral vaccine is 1 in 2.5 million. (6) Mortality is 5–10%. Motor strength usually returns in 4 months. b. Coxsackie and echoviruses—cause meningoencephalitis and polymyositis. They may be cultured from the CSF. The incidence is increased in patients with humoral immunity deficiencies. 15. Mumps—10% of the patients with mumps parotiditis develop meningitis. Before the routine use of the mumps vaccine, it accounted for 25% of viral encephalitis cases. It can be cultured from the CSF. 16. Measles—a postinfectious encephalomyelitis develops after the rash in 1 in 1000 cases. It is immune mediated and not directly caused by the virus. a. SSPE—occurs in children and adolescents (ages 5–15 years) after a measles infection that has usually occurred before 2 years of age. b. Chronic encephalitis develops with deteriorating school performance and behavioral changes followed by myoclonus, seizures, weakness, and death in 1–3 years. c. It affects both the gray and white matter with perivascular lymphocytes, neuronophagia, demyelination, and oligodendrocyte destruction. d. There are intranuclear and intracytoplasmic inclusions in the neurons and oligodendrocytes. The CSF has oligoclonal antibodies to the measles virus, no cells, and increased protein (Fig. 3.42). e. Diagnosis can be made by periodic 2–3 electroencephalographic (EEG) spikes per second, increased CSF immunoglobulin, and increased serum and CSF measles antibody titers. Fig. 3.42 Subacute sclerosing panencephalitis (hematoxylin and eosin [H&E] stain); eosinophilic intranuclear and intracytoplasmic inclusions (arrow). a. The JC virus normally resides in the kidney. PML occurs in immunocompromised people and is found in 2% of AIDS autopsies. b. Typically, CT and MRI demonstrate patchy hypodense white matter changes without enhancement or mass effect (Fig. 3.43). c. It is bilateral, asymmetric, subcortical, spares the cortex, and usually starts in the posterior centrum semiovale. There is minimal cellular infiltration, central destruction of oligodendrocytes, demyelination, and peripheral swollen irregular oligodendrocytes with ground-glass nuclei and intranuclear inclusions of “stick-and-ball” viral particles. CSF is normal (Fig. 3.44 and Fig. 3.45). d. Death occurs within a few months. Fig. 3.43 Progressive multifocal leukoencephalopathy. (A) Computed tomographic, (B) proton density, and (C) T2-weighted magnetic resonance images demonstrating patchy white matter degeneration. Fig. 3.44 Progressive multifocal leukoencephalopathy (hematoxylin and eosin [H&E] stain); low-power view demonstrates demyelination in cerebellar sub-cortical white matter. Fig. 3.45 Progressive multifocal leukoencephalopathy (hematoxylin and eosin [H&E] stain). High-power view demonstrates demyelination with surrounding enlarged bizarre oligodendrocyte nuclei (arrows). 18. Transmissible spongiform encephalitis—The incubation is months to years, but once deterioration is started it progresses rapidly to death. These are a heterogeneous collection of probable infections caused by SSPE and HIV, but most commonly prions. 19. Prions—These are not viruses because they are protein without nucleic acid. They are resistant to nucleases, ultraviolet light, and radiation. They can be inactivated with autoclaving for 1 hour at 132°C or immersion in 1N NaOH for 15 minutes or 0.5% sodium hypochlorite for 1 hour. a. A protease-resistant protein is coded on chromosome 20 and the production may be released by a DNA configuration change. b. Prion diseases—kuru (in cannibals of New Guinea), scrapie (in sheep), chronic wasting disease in elk and mink, fatal familial insomnia, Creutzfeldt–Jakob disease (CJD), and Gerstmann–Sträussler syndrome. CJD occurs in 50- to 65-year-old people and manifests as myoclonus, pyramidal, and extrapyramidal degeneration, dementia, ataxia, and visual deterioration. c. There is a characteristic EEG with bilateral sharp waves of 1–2 waves/s that resemble periodic lateralized epileptiform discharges, but are reactive to painful stimuli. d. May have positive CSF immunoassay for 14-3-3 protein; fairly sensitive and specific e. Pathologic examination—demonstrates spongiform changes with astrocytosis but no inflammation most prominently in the cortex, putamen, and thalamus (Fig. 3.46). 5–10% of patients develop amyloid kuru plaques. Death occurs in less than 1 year. f. Gerstmann–Sträussler syndrome—ataxia, dysarthria, hyporeflexia, and cognitive decline, without myoclonus. Transmission is both autosomal dominant and sporadic, and Kuru plaques are seen in the cerebellum. These diseases can be transmitted to laboratory animals that develop the same microscopic changes. g. Fatal familial insomnia—sleep disturbance, agitation, mild cognitive changes, and dysautonomia; characterized by thalamic atrophy 20. Acquired immune deficiency syndrome a. Acquired immune deficiency syndrome (AIDS)—caused by a retrovirus that contains RNA. It requires reverse transcriptase to convert its RNA to DNA and allow replication. b. HTLV-1 (human T cell lymphotropic virus)—causes a chronic myelopathy with spastic paresis, but no sensory deficit and involves the anterior and lateral columns. It occurs mainly in the Tropics (tropical spastic paraparesis) and Japan. Fig. 3.46 Creutzfeldt–Jakob disease (hematoxylin and eosin [H&E] stain); spongiform changes. c. HTLV-2—associated with chronic T cell leukemia and lymphoma d. HIV (HTLV-3)—a lentivirus with a surface molecule that interacts with T4. It is associated with Pneumocystis infections and Kaposi sarcoma. Viral latency is 8 years, and once AIDS ensues, 50% die in <1 year and most in <3 years if untreated. 50% of cases develop neurologic symptoms and 80% have CNS abnormalities. CNS complications at various stages include (1) Seroconversion—aseptic meningitis, acute encephalitis, and myelopathy (2) HIV-positive—asymptomatic or AIDS-related complex with aseptic meningitis (3) AIDS—aseptic meningitis, AIDS dementia complex, lymphoma, and vacuolar myelopathy e. AIDS dementia complex—the most frequent AIDS disorder seen in 50% of patients. It is believed to be caused by encephalitis or the viral impact on the neurons. It consists of a triad of cognitive dysfunction (subcortical dementia), behavioral changes (psychoses and hallucinations), and motor deficits (by centrum semiovale and brainstem involvement). f. Congenital lesions—atrophy, hydrocephalus ex vacuo, microcephaly, facial dysmorphism, and basal ganglia calcifications g. HIV encephalitis—pathologic examination demonstrates microglial nodules in the white matter and subcortical gray matter with focal demyelination, neuronal loss, reactive astrocytosis, and calcifications in the parenchymal blood vessels and basal ganglia. There are characteristic multinucleated giant cells that are unique to HIV in the CNS and are of macrophage origin (Fig. 3.47). h. Vacuolar myelopathy—mainly involves the posterior and lateral columns of the low thoracic levels. Pathologic examination demonstrates vacuolar changes with lipid-laden macrophages. It is detected in 50% of AIDS autopsies, but less often clinically. i. Peripheral neuropathy—seen in 5–38% of AIDS cases and is caused by didanosine, demyelination, and arteritis j. Myopathy—seen in 20% of AIDS cases, is characterized by inflammation, atrophy, polymyositis, and azathioprine-induced mitochondrial changes k. Toxoplasmosis—seen in 10% of autopsy cases. It is the most frequent cause of focal neurologic symptoms in AIDS. Toxoplasmosis may cause focal or diffuse lesions and predominantly causes basal ganglia and gray/white matter junction lesions. The serum IgG level is increased with infection, and pathologic examination demonstrates a mononuclear inflammation with extracellular tachyzoites and encysted bradyzoites (Fig. 3.37). Treatment is with pyrimethamine and sulfadiazine for 14 days. Fig. 3.47 Human immunodeficiency virus encephalitis (luxol fast blue-hematoxylin and eosin [H&E] stain). Microglial nodule with characteristic multinucleated giant cells. l. CMV—seen in 30% of AIDS autopsies, but only 10% are symptomatic with necrotizing encephalomyelitis. Pathologic examination demonstrates microglial nodules, mainly in the gray matter and rarely in the white matter (unlike HIV). There are Cowdry type A intranuclear and intracytoplasmic inclusions in the neurons and astrocytes. m. PML—described above. n. Primary CNS lymphoma—seen in 5% of autopsies. It is invariably of B cell origin with large or mixed large/small cell types. They are usually periventricular with perivascular spread. Epstein–Barr virus (EBV) is frequently detected in the cells and may play a role in the development of the lymphoma. o. Cryptococcus—in the normal population it causes meningitis, whereas in immunocompromised patients it causes encephalitis and cryptococcomas in the basal ganglia and midbrain p. AIDS symptomatic infections—HIV encephalitis (60%), toxoplasma (30%), Cryptococcus (5%), PML (4%), and less frequently lymphoma (not an infection), TB, syphilis, varicella-zoster, and CMV 21. Rasmussen chronic encephalitis—occurs in childhood and causes progressive deficits and seizures (classically complex partial status epilepticus) with unilateral atrophy. It may be caused by CMV infection or antibodies to glutamate receptors. 22. Postinfectious encephalitis a. SSPE—It develops several years after a measles infection. There are increased neutralized measles virus immunoglobulin titers in the serum and CSF. Pathologic examination demonstrates atrophy, perivascular lymphocytosis, demyelination, and increased eosinophilic intranuclear and intracytoplasmic inclusions in the neurons and oligodendrocytes. It affects children and young adults and develops in 1 in 1,000,000 cases of measles. Findings are as described above. Death occurs within 1–3 years. b. Acute disseminated encephalomyelitis—immune-mediated disease that occurs after a viral infection or a vaccination (1) It most commonly occurs after varicella-zoster, measles, upper respiratory infection, rabies vaccination, diphtheria, smallpox, tetanus, typhoid, and influenza. (2) Pathologic examination demonstrates perivascular mononuclear infiltrates with a zone of demyelination along the course of the venules. Congenital Infections (Torch) 219 (3) All ages are affected, but it usually occurs in children and young adults. (4) Symptoms occur 1–3 weeks after the infection. It follows a rapid course, although some patients survive. (5) Treatment is with steroids. Mortality is 20–50%. 23. CSF glucose—low with fungal infections, TB, carcinomatous meningitis, and sarcoid. 24. Interleukin-1—released in response to endotoxins and stimulates the interleukin cascade to increase T cell proliferation 25. Tumor necrosis factor—stimulates interleukin-1 production and activates neutrophils A. Congenital infections are acquired three ways: 1. Hematogenous spread through the placenta (toxoplasma and viruses) 2. Ascending from the cervix (bacteria) 3. During passage through the birth canal (herpes, this mechanism causes neonatal rather than true congenital infection) 4. They may cause developmental changes or tissue destruction. Larger organisms such as bacteria, fungus, and protozoa are unable to enter the embryo before 3 to 4 months, but viruses may pass through the placenta. 5. The usual time frame for congenital infections is first trimester (rubella), 4 months (syphilis), 5 months (toxoplasma), perinatal (bacteria, HIV), during passage through birth canal (HSV-2, HIV). B. Cytomegalovirus 1. CMV is the most frequent congenital CNS infection. 2. It causes migration disorders during the first or early second trimester. Transmission is transplacental. 75% of women have serum CMV antibodies (which are protective), 1% of newborns have CMV detected in the urine, and 10% of these have CNS infection. 3. It affects the brain, heart, liver, and spleen. The virus has an affinity for the germinal matrix and causes perivascular necrosis and calcifications. 4. Premature infants may have hepatosplenomegaly, jaundice, thrombocytopenia, chorioretinitis, seizures, mental retardation, optic atrophy, impaired hearing, and hydrocephalus. 5. Diagnosis is made by cultures, immunoglobulin levels, and intracellular (intranuclear and intracytoplasmic) inclusions on biopsy specimens. 6. Radiographs may demonstrate microcephaly and periventricular eggshell calcifications. C. Toxoplasma 1. Toxoplasma is the second most frequent congenital CNS infection. 2. It occurs in 1 in 1000 to 1 in 10,000 pregnancies and is acquired by hematogenous spread through the placenta. There may be giant cell granulomas with atrophy of the basal ganglia, periventricular white matter, and cortex. 4. Findings include seizures, microcephaly, and spontaneous abortions. A classical triad includes hydro-cephalus, bilateral chorioretinitis, and cranial calcifications. Deafness is the most common late manifestation. 5. Treat with spiramycin if the mother seroconverts in 2–3 months. If the mother seroconverts after 4 months, use pyrimethamine and sulfadiazine. D. Rubella 1. Rubella is transmitted transplacentally. 2. The virus inhibits cell multiplication to cause an insufficient number of cells in the brain (less neurons, astrocytes, and oligodendrocytes) and also is teratogenic and destructive. 3. The infection may cause meningoencephalitis, vasculopathy with ischemia and necrosis, microcephaly, decreased myelin, and cortical and basal ganglia calcifications. If the infection occurs before 12 weeks, the effects are very severe and spontaneous abortion is likely. If it occurs after 12 weeks, the infection is less severe. 4. Congenital rubella syndrome occurs with infection in the first trimester and includes chorioretinitis, cataracts, glaucoma, microphthalmia, microcephaly, mental retardation, and deafness. 5. During infection, the CSF has increased mononuclear cells and IgM; prevent with maternal vaccination before pregnancy. E. Herpes simplex virus 1. HSV is transmitted transvaginally. 2. It occurs in 1 in 200 to 1 in 5000 births. 85% are caused by HSV-2 and rare early lesions cause death, chorioretinitis, and microcephaly. The virus has a predilection for vascular endothelial cells and causes thrombosis and hemorrhagic stroke that develops 2–4 weeks postpartum. 3. Pathologic examination demonstrates microglial nodules and intranuclear inclusions. The infection is diffuse and causes white matter edema without the temporal localization. 4. The most frequent manifestations are skin, eye, and mouth lesions, but if no treatment is given, the infection may disseminate with an 80% mortality (only 50% with treatment). 5. The CNS is involved in 30% and produces fever, seizures, and lethargy. HSV-1 affects older children. F. Human immunodeficiency virus 1. HIV can be transmitted perinatally. 30% of HIV mothers transmit the virus to the children. 2. Pathologic examination demonstrates brain atrophy and basal ganglia calcifications. 3. Symptoms include weight loss, failure to thrive, diarrhea, and fever. Most die in 1 year. G. Syphilis 1. Syphilis has transplacental transmission at 4–7 months. 2. Hutchinson triad—dental disorders, bilateral deafness, and interstitial keratitis 4. The child should be treated with penicillin until the CSF is acellular and has a normal protein. H. Listeria—causes increased abortions and premature deliveries I. Calcifications—caused mainly by CMV (periventricular) and toxoplasma (disseminated) J. Cardiac malformations—caused by rubella K. Deafness—caused by rubella, syphilis, and CMV A. General Neurooncology 1. Primary brain tumors are less common than metastatic tumors. The most frequent is glioblastoma multiforme (GBM) followed by meningioma. Gliomas are more common in males, and meningiomas are more common in females. 2. Children—Brain malignancies are the second most common cancer after leukemia. 70% are infratentorial. The most frequent types are cerebellar astrocytoma (33%), brainstem glioma (25%), medulloblastoma (25%), and ependymoma (12%). 20% of brain tumors occur before 15 years of age. The most frequent supratentorial tumors in children are low-grade astrocytomas (50%), craniopharyngiomas (12%), and optic gliomas (12%). 3. Neonates and infants (<2 years)—Brain tumors are rare and usually congenital. Two thirds are supratentorial. The most common is teratoma, followed by primitive neuroectodermal tumors (PNETs), high-grade astrocytoma, and choroid plexus papilloma. Findings include macrocephaly, hydrocephalus, split sutures, seizures, and focal deficits. Most of the tumors are highly malignant and have a poor prognosis. 4. Older children—Most brain tumors are infratentorial. The most common types are astrocytoma (50%), PNET (15%), craniopharyngioma (10%), ependymoma (10%), and pineal tumor (3%). 5. Adults—Primary tumors are less common than metastatic tumors in clinical series. 70% are supratentorial. The most frequent tumors are GBM, metastases, anaplastic astrocytoma, meningioma, pituitary tumors, and vestibular schwannomas. The most frequent infratentorial tumors are metastases, schwannoma, meningioma, epidermoid, hemangioblastoma (the most frequent primary intraaxial posterior fossa tumor), and brainstem glioma. 6. In the spinal cord—The most frequent epidural lesions are metastatic. The most frequent intradural/extramedullary lesions are schwannoma and meningioma. The most frequent intramedullary lesions are astrocytoma and ependymoma. 7. Causes of tumors a. Radiation—associated with meningiomas, fibrosarcomas, and gliomas b. Immunosuppression—associated with lymphomas c. Viruses—EBV is associated with Burkitt lymphoma and nasopharyngeal carcinoma, and the human papillomavirus is associated with cervical carcinoma. d. Chemotherapy—nitrosoureas e. Genetics—as seen in phakomatoses and Turcot syndrome (APC gene mutation on chromosome 5q with familial polyposis, colorectal cancer, and primary brain tumors) 9. Analysis of tumors—light microscopy, electron microscopy, immunohistochemistry, and in situ messenger RNA hybridization 10. Immunohistochemical stains a. AFP—embryonal carcinoma, endodermal sinus tumor b. CEA – carcinoembryonic antigen c. Chromogranin—pituitary adenoma d. Common leukocyte antigen—lymphoma, germinoma e. Cytokeratin—carcinoma, craniopharyngioma, chordoma f. Desmin—rhabdosarcoma, teratoma g. Epithelial membrane antigen (EMA)—carcinoma, meningioma, epithelial cysts h. GFAP—astrocytomas, other glial tumors i. Human melanoma black (HMB)—melanoma j. Beta human chorionic gonadotrophin (HCG)—choriocarcinoma and the syncytiotrophoblastic variant of germinomas k. Immunoglobulins kappa and lambda chains—lymphomas l. Neurofilament and synaptophysin—ganglioglioma, PNET m. Pituitary hormones—pituitary adenoma n. Prostate specific antigen—prostate carcinoma o. S100—schwannoma, neurofibroma, glioma, PNET, chordoma, melanoma, renal cell carcinoma p. Synaptophysin—tumors with neurons (ganglioglioma, central neurocytoma, etc.) q. Transthyretin—choroid plexus tumors r. Vimentin—meningioma s. Many tumors have unexpected overlaps. 11. Assessment of proliferative capacity a. G1 phase—preparation for DNA synthesis (susceptible to radiation) b. S phase—DNA synthesis (resistant to radiation) c. G2 phase—preparation for mitosis d. M phase—mitosis (susceptible to radiation) e. G0 phase—nondividing quiet phase of cell cycle 12. Only a small portion of the dividing cells is in the M phase, so the mitotic index is misleading. Flow cytometry is more accurate because it stains DNA and counts the number of cells with double DNA. Also available are cytophotometry, DNA synthesis markers, and proliferation antigens that appear during the cell cycle (Ki67 is expressed in all stages except G0). 13. Oncogenes a. Oncogene—overexpression of an oncogene can cause the cell to enter an unrestrained replication cycle (malignant change). They may be introduced by a virus. Examples are c-myc oncogene (activation causes Burkitt lymphoma) and n-myc oncogene 9 (activation causes neuroblastoma). b. Proto-oncogene—a gene locus that becomes an oncogene by deletion or translocation c. Tumor suppressor gene—a gene in the normal genome that when lost or mutated allows malignant growth to occur. Tumor suppressor gene loss is associated with gliomas (chromosomes 9, 10, and 17), meningiomas (chromosome 22), and retinoblastomas (chromosome 13). The p53 nuclear protein gene is a tumor suppressor gene on chromosome 17p, and alterations are seen in 33% of astrocytomas. 14. Paraneoplastic syndromes (possibly autoimmune or viral) a. Limbic encephalitis—subacute encephalitis. Gross examination appears normal. There are perivascular mononuclear infiltrates but no viral inclusions. The medial temporal lobes, cingulate gyrus, and insula are predominantly affected. There are usually bilateral hyperintense lesions on T2-weighted MRI. It is most common in men in their mid-60s and manifests as memory impairment and altered mental status. It may be associated with testicular or lung cancer and anti-Ma protein antibodies. Limbic encephalitis should be differentiated from herpes encephalitis. b. Anti-Yo antibodies—cause cerebellar degeneration and are associated with ovarian and breast cancer c. Anti-Hu antibodies—cause sensory neuropathy, encephalitis, and cerebellar degeneration. They are associated with oat cell pulmonary carcinoma or lymphoma. d. Anti-Ri antibodies—cause opsoclonus and are associated with breast cancer e. Eaton–Lambert syndrome—antibodies to the presynaptic voltage-gated Ca2+ channels; associated with oat cell (small cell) lung carcinoma. See section XXXIII. f. Stiff man syndrome—involuntary muscle spasms and rigidity; 60% have antibodies to glutamic acid decarboxylase 15. Radiation a. Radiation-sensitive tumors—Lymphoma and germ cell tumors (mainly germinoma but to some extent the others) are very radiosensitive. Meningioma, pineal tumors, craniopharyngioma, pituitary tumors, vestibular schwannoma, and metastatic tumors are less sensitive. b. Standard radiation doses (1) Metastatic tumors—30 Gray (Gy) over 2 weeks (2) Gliomas—6000 centigray (cGy) in 200 cGy daily fractions (3) Doses higher than these are much more likely to cause radiation necrosis. c. Radiation necrosis—white matter coagulation necrosis or demyelination associated with arterioles with hyalin intimal thickening, fibrinoid necrosis, and thrombosis. The neurons are relatively resistant. Symptoms usually start 3 months–3 years (average 15–18 months) after radiation. Radiation myelopathy is reduced if the daily fraction is kept less than 200 cGy, the weekly fraction less than 900 cGy, and the total dose less than 6000 cGy. Symptoms begin with paresthesias/dysesthesias in the hands or feet and Lhermitte sign. There is no local pain, and it may progress irregularly. T2-weighted MRI demonstrates increased signal intensity, and pathologic examination demonstrates necrosis of the gray and white matter. There is no effective treatment: steroids are sometimes tried (Fig. 3.48 and Fig. 3.49). d. Radiation may induce tumor formation such as sarcomas, GBMs, and meningiomas. B. Glial tumors 1. Astrocyte types are a. Fibrillary—more numerous, mainly in the white matter, stains with phosphotungstic acid hematoxylin (PTAH), silver, and GFAP b. Protoplasmic—mainly in the gray matter and has a larger nucleus, but less cytoplasm c. Gemistocytic—swollen active astrocyte with increased fibers and cytoplasm and appears often with injury, stroke, toxin, infection, or tumor 2. Circumscribed astrocytic tumors—low grade, good prognosis, and frequently cystic a. Juvenile pilocytic astrocytoma (grade 1 astrocytoma)—the second most common pediatric brain tumor. It accounts for one-third of pediatric gliomas and 5 to 10% of all gliomas. It is most frequently located in the cerebellum, brainstem, optic pathway, and infundibulum. In adults, it is more common near the third ventricle. The peak age is 10 years. 60% are cystic and they usually have a mural red-tan nodule. There is a biphasic pattern of loose cells and micro-cysts and also dense elongated hair-like astrocytes with Rosenthal fibers and eosinophilic granular bodies. They tend to be noncystic in the medulla and optic pathway. Leptomeningeal invasion, nuclear atypia, multi nucleated cells, and vascular proliferation are frequently noted but are not adverse prognostic indicators. Approximately 10% contain calcium. The nodule may enhance. There is usually no necrosis. Survival rate is 86–100% at 5 years, 83% at 10 years, and 70% at 20 years (Figs. 3.50 – 3.54). Fig. 3.48 Radiation necrosis. (A) Nonenhanced and (B) enhanced axial T1-weighted magnetic resonance images demonstrate the irregularly enhancing low-density lesion. Fig. 3.49 Vertebral body radiation changes. Sagittal T1-weighted magnetic resonance image demonstrates the high-signal intensity changes in the vertebral bodies because of increased fat content. Fig. 3.50 Normal gray matter (hematoxylin and eosin [H&E] stain). Neurons oriented with large apical dendrites (double arrows) toward the pia and axons (arrows) toward the ventricles. Fig. 3.51 Normal white matter (hematoxylin and eosin [H&E] stain). Normal oligodendrocytes interspersed in a fibrillary background. Fig. 3.52 Juvenile pilocytic astrocytoma. Axial T1-weighted magnetic resonance image (MRI). (A) nonenhanced and (B) enhanced, and sagittal T1-weighted (C) nonenhanced and (D) enhanced MRI scans with an enhancing nodule in a cerebellar cystic lesion. Fig. 3.53 Pilocytic astrocytoma (hematoxylin and eosin [H&E] stain). (A) Biphasic histologic pattern of a loose microcystic component and a dense component with higher-power views of the (B) loose and (C) dense areas. Fig. 3.54 Pilocytic astrocytoma (hematoxylin and eosin [H&E] stain); eosinophilic Rosenthal fibers. c. Subependymal giant cell astrocytoma—the peak age is <20 years. Symptoms are by hydro-cephalus and seizures. They are located near the foramen of Monro. They enhance, have frequent calcifications, may be cystic and lobulated, and tend to be well demarcated. Pathologic examination reveals large multinucleated cells and rare mitoses. These tumors are seen in 15% of patients with tuberous sclerosis, and if found in a patient without tuberous sclerosis, it is considered a forme-fruste (Fig. 3.55 and Fig. 3.56). 3. Diffuse astrocytic tumors—infiltrative and carry a worse prognosis. They may be fibrillary (most frequent), protoplasmic, gemistocytic (probably a worse prognosis if > 20% of cells are gemistocytic) or mixed. The grade is determined by the degree of anaplasia with increased cellularity, nuclear pleomorphism, mitoses, endothelial proliferation, necrosis, and to a lesser extent, pseudopalisading features. Fig. 3.55 Subependymal giant cell astrocytoma. (A) Axial nonenhanced and (B) enhanced computed tomo-graphic scans demonstrating a large enhancing mass in the lateral ventricle. Also noted is a calcified tuber on the right lateral ventricular wall. Fig. 3.56 Subependymal giant cell astrocytoma (hematoxylin and eosin [H&E] stain). Enlarged cells with abundant eosinophilic cytoplasm and large nuclei with prominent central nucleoli. a. Grade II (low-grade astrocytoma)—well differentiated, represent 15% of astrocytomas, peak age is 30 years, male predominance, most frequently in the frontal white matter and most com monly fibrillary. They tend to be hypodense, minimally enhancing, and occasionally cystic. Pathologic examination reveals a gray-colored homogeneous tumor with indistinct borders that expands the white matter. There are no mitoses and only rarely hemorrhage or edema. 15% calcify. It is differentiated from reactive gliosis because it is patternless, violates the gray/white matter junction, has microcysts, microcalcifications, and increased nuclear atypia and pleomorphism. The 5-year survival rate with total resection and radiation is 70% and with subtotal resection and radiation is 38%. Median survival rate is 8.2 years and 50% increase in grade over time (Fig. 3.57 and Fig. 3.58). Fig. 3.57 Low-grade astrocytoma. Axial computed tomography scans (A) nonenhanced and (B) enhanced with nonenhancing left frontal hypo-dense mass. Fig. 3.58 Fibrillary (grade 2) astrocytoma (hematoxylin and eosin [H&E] stain). Patternless but uniform infiltrating cells. b. Grade III (anaplastic astrocytoma)—represent 30% of astrocytomas; peak age is 40–60 years, with male predominance. CT reveals a mixed density lesion with irregular rim enhancement and edema. Pathologic examination demonstrates increased cellularity, nuclear atypia, mitotic figures, with or without endothelial hyperplasia, and no necrosis. There may be hemorrhages or cysts and rarely calcification. There are frequent gemistocytes (>20 per high power field denotes worse prognosis). They spread through white matter tracts, and there is frequent ependymal and CSF dissemination. Secondary structures of Scherer are located around neurons in the gray matter, the subpial region, and the subependymal zone. The median survival rate is 2–3 years (Fig. 3.59). c. Grade IV (GBM)—represent 50% of astrocytomas, the most frequent primary brain tumor (20%), peak age is 45 to 60 years, with male predominance. GBM and anaplastic astrocytoma are among the four most common CNS tumors. (1) The location is most commonly deep frontotemporal. CT reveals a heterogeneous lesion that is cystic in 85% of cases with rare calcifications. Angiogram demonstrates a vascular mass with arteriovenous shunting and early draining veins (Fig. 3.60). Fig. 3.59 Anaplastic (grade 3) astrocytoma (hematoxylin and eosin [H&E] stain). Increased cellularity with hyper-chromatic, pleomorphic nuclei. Fig. 3.60 High-grade astrocytoma. (A) Axial nonenhanced and (B) enhanced T1-weighted magnetic resonance images demonstrating a temporal lesion with irregular cystic enhancement characteristic of a glioblastoma multiforme. (2) Pathologic examination is heterogeneous with cysts, degeneration, necrosis, hemorrhages, edema, marked hypercellularity, nuclear atypia, frequent mitoses, pseudopalisading, and endothelial hyperplasia with glomeruloid structures (Fig. 3.61 and Fig. 3.62). (3) The tumor cells follow white matter tracts (especially the corpus callosum) and may invade dura, disseminate in the CSF, and occasionally produce distant metastases. They are occasionally multicentric (3–6%). GBMs are associated with increased epithelial growth factor receptor (on chromosome 7). The median survival rate is 9–12 months. (4) Giant cell GBM—has multinucleated giant cells, increased reticulin, and a slightly better prognosis probably because the giant cells can no longer divide. Small cell GBM has a worse prognosis. (5) Gliomatosis cerebri—one or two diffusely enlarged hemispheres filled with tumor or diffuse enlargement of the cerebellum or brainstem. It is most common at 20–30 years, and there are no focal masses (Fig. 3.63). Fig. 3.61 Glioblastoma multiforme (hematoxylin and eosin [H&E] stain); pseudopalisading necrosis. Fig. 3.62 Glioblastoma multiforme (hematoxylin and eosin [H&E] stain); endothelial proliferation. Fig. 3.63 Gliomatosis cerebri. Coronal (A) T1-weighted, (B) axial proton density, and (C) T2-weighted magnetic resonance images demonstrating diffuse bilateral white matter infiltration by the tumor. (6) In addition to maximal surgical resection and external beam radiation, the concomitant use of temozolomide chemotherapy during and after radiation therapy significantly prolongs survival compared with radiotherapy alone in patients with newly diagnosed GBM – representing a new standard of care. Epigenetic silencing (DNA methylation) of the O6-methylgua-nine-DNA methyltransferase (MGMT) gene, which encodes a DNA repair protein and represents an important mechanism of chemotherapy resistance, is an independent predictor of improved survival as well as survival benefit from temozolomide. d. Protoplasmic astrocytoma—small stellate cells with delicate processes that are predominantly in the gray matter. Prognosis is similar to fibrillary astrocytomas. e. Adult pilocytic astrocytoma—unlike the juvenile variety. It is not circumscribed and has a worse prognosis. f. Gemistocytic astrocytoma—defined by >20% gemistocytes, contains large cells with eccentric eosinophilic cytoplasm, and has a worse prognosis (Fig. 3.64). Fig. 3.64 Gemistocytic astrocytoma (hematoxylin and eosin [H&E] stain). Enlarged astrocytes with prominent eosinophilic cytoplasm and eccentric nuclei. g. Gliosarcoma—represents 2% of GBMs, peak age is 40–60 years. It most commonly involves the temporal lobe superficially with dural invasion. Lesions are firm, circumscribed, lobulated, and contain fascicles of spindle cell sarcoma with interspersed GBM cells. Silver stains the reticulin in the sarcoma component and GFAP stains the GBM component. There are frequent intracranial and extra-cranial metastases (15–30%). It is postulated that the sarcoma arises from the vascular structures in the GBM or from leptomeningeal fibroblasts. The survival rate is similar to GBM (Fig. 3.65). Fig. 3.65 Gliosarcoma (hematoxylin and eosin [H&E] stain). Biphasic pattern of neoplastic glial and mesenchymal cells with reticulin background. h. Optic glioma—peak age is 3–5 years, female predominance, 20% may act malignantly, associated with neurofibromatosis type I (NF1). Treatment is (1) if distal to the chiasm, remove optic nerve and attached globe; and (2) if the chiasm is involved, resect up to the chiasm preserving vision in the better eye and consider radiation if tumor progression is noted (Figs. 3.66 – 3.68). i. Brainstem glioma—accounts for 20% of intracranial tumors in children, usually diffusely infiltrates and enlarges the pons, progresses rapidly. The 5-year survival rate is 30%. Symptoms begin with cranial nerve palsies, and hydro-cephalus develops late. Treatment is with radiation. The one tumor for which biopsy is usually not necessary because the diffuse pontine lesion (hypointense on T1-weighted MRI and nonenhancing) is very characteristic. The prognosis is better with cystic lesions, dorsal exophytic lesions, and lesions involving the midbrain, medulla, or cervicomedullary junction. These lesions may be amenable to surgical resection (Fig. 3.69). Fig. 3.66 Optic glioma. Nonenhanced axial computed tomographic scan demonstrating thickened right optic nerve (arrow). (From Valvassori GE, Mafee MF, Carter BL. Imaging of the Head and Neck. New York, NY: Thieme; 1995. Reprinted by permission.) Fig. 3.67 Optic chiasm glioma. (A) T1-weighted nonenhanced sagittal and (B) enhanced coronal magnetic resonance images demonstrating an expansile enhancing lesion of the optic chiasm. Fig. 3.68 Optic chiasm glioma. Diffuse enlargement with extension under chiasm. 4. Oligodendroglioma a. Accounts for 10% of gliomas, peak age incidence is 35–40 years, and there is no sex predominance. b. Oligodendrogliomas can be pure or mixed with astrocytic components (i.e., oligoastrocytoma). Histological grade II is pure or mixed and histological grade III is anaplastic. c. Symptoms frequently include seizures. They grow from the white matter and infiltrate the cortex. CT demonstrates a hypodense lesion, which is frequently cystic. It has a higher frequency of hemorrhage than other glial tumors (Fig. 3.70). d. Pathologic examination demonstrates round nuclei with scant cytoplasm, a chicken-wire vascular pattern with thin vessels, occasional serpentine configuration, and an Indian-file lineup of cells in the white matter with satellitosis of neurons in the gray matter. The fried egg yolk–appearing cells and nucleus are caused by an artifact from cytoplasmic retraction seen in permanent, but not in frozen sections. 80% have calcifications. Immunohistochemistry is positive for GFAP and S100 (Fig. 3.71). Fig. 3.69 Pontine glioma. (A) T1-weighted sagittal, (B) nonenhanced and (C) enhanced axial, and (D) T2-weighted axial magnetic resonance images with low-intensity nonenhancing expansion of the pons. Fig. 3.70 Oligodendroglioma. (A) T1-weighted axial enhanced and (B) T2-weighted coronal magnetic resonance images demonstrating right posterior frontal hemorrhagic tumor. (From Yasargil MG. Microneurosurgery IV A. New York, NY: Thieme; 1994.) e. Systemic metastases are rare and usually occur after surgery. The 5-year survival rate is better than for astrocytomas. The most important prognostic factor is grade (1–4). The low-grade tumors have a 5-year survival rate of 74% and 10-year survival rate of 46%. The high-grade tumors have a 5-year survival rate of 41% and a 10-year survival rate of 20%. Grades 3 and 4 have endovascular proliferation and hypercellularity. PCV (procarbazine, carmustine, and vincristine) or temozolomide chemotherapy is used for oligodendrogliomas. Fig. 3.71 Oligodendroglioma (hematoxylin and eosin [H&E] stain). Uniform “fried-egg” cells with geometric “chicken-wire” arrangement of vessels. f. Combined loss of heterozygosity of chromosomes 1p and 19q (the most common genetic alteration in oligodendrogliomas) is associated with a better response to chemotherapy and improved progression-free and overall survival in patients with anaplastic oligodendrogliomas. 5. Ependymoma a. Peak age is 10–15 years, with a large peak at 1–5 years and smaller peak at 35 years. There is no sex predominance. b. Location is usually infratentorial. They may grow from the fourth ventricle out through the foramen of Luschka and Magendie. They also account for 60% of intramedullary spinal cord tumors, occurring mostly at the filum. c. They are usually pencil-shaped in the spinal cord, often associated with a syrinx, and have a good margin for resection. There may be multiple spinal cord tumors with neurofibromatosis type 2 (NF2). The myxopapillary variety occurs only at the filum (normally it has a good prognosis, but is worse if it invades the conus). d. CT and MRI demonstrate a lobulated, circumscribed, cystic, moderately enhancing lesion with calcifications (50%) and only rarely hemorrhage. Grossly it is tan-red in color (Fig. 3.72). e. Pathologic examination demonstrates various patterns: (1) Cellular—a sheetlike growth of polygonal cells with true rosettes (around a central canal), pseudorosettes (around a blood vessel), and blepharoplasts (ciliary basal bodies in the apical cytoplasm) (Fig. 3.73). (2) Papillary—with typical papillary projections (3) Myxopapillary—with intracellular mucin, occurs at the filum and presacral/postsacral area if there is local spread (Fig. 3.74) (4) Clear cell—with oligodendrocyte-like halos f. Immunohistochemistry is positive for GFAP and histochemistry for PTAH. These can be differentiated from (1) medulloblastoma by the smaller nuclei, fewer mitoses, absence of Homer Wright rosettes, positive GFAP, and negative synaptophysin; and (2) choroid plexus papilloma that are PTAH negative and cytokeratin positive. Fig. 3.72 (A) Ependymoma. T1-weighted axial nonenhanced and (B) coronal enhanced magnetic resonance images demonstrating an enhancing fourth ventricular mass extending through the foramen of Magendie and Luschka. Fig. 3.73 Ependymoma (hematoxylin and eosin [H&E] stain); pseudorosette (around a vessel). Fig. 3.74 Myxopapillary ependymoma (hematoxylin and eosin [H&E] stain). Cohesive ependymal cells terminating around mucin-rich perivascular spaces. h. Ependymoblastoma (grade IV) occurs in childhood, is in the PNET group, and is malignant. i. The normal ependyma consists of a single layer of cuboidal/columnar cells that are ciliated early in life and have microvilli. They have a dual epithelial–glial nature and lie over the subependymal glia. 6. Subependymoma a. Peak age is 40–60 years, with male predominance and usually located in the floor of the inferior fourth ventricle or the lateral ventricle. b. They arise at the ependymal–subependymal zone and grow slowly. They are benign, avascular, nonenhancing, firm, well circumscribed, hypocellular, and nodular with rests of cells separated by glial fibrils (Fig. 3.75 and Fig. 3.76). Fig. 3.75 Subependymoma. (A) Nonenhanced and (B) enhanced T1-weighted axial magnetic resonance images demonstrating a pedunculated nonenhancing lateral ventricular mass (the most common location is the fourth ventricle). Fig. 3.76 Subependymoma (hematoxylin and eosin [H&E] stain). Hypo-cellular with small nests of cells separated by broad bands of fibrils d. Only 50% become symptomatic, usually by CSF obstruction. A gross total resection can usually be achieved except at the floor of the fourth ventricle. It is postulated that this may be a form of ependymoma because they are often mixed. 7. Choroid plexus papilloma a. Represents <1% of brain tumors and the peak age is >10 years. It is one of the most frequent tumors before 2 years. b. 50% are located in the lateral ventricle (more commonly left atrium in children), 40% are in the fourth ventricle (more commonly in adults), 10% is in the third ventricle. Rarely, they occur in the CPA. 4% are bilateral. c. They tend to be well circumscribed, vascular, and enhancing. They have a cauliflower papillary shape with cuboidal and columnar cells and no cilia (except in children). The cells are piled up on stalks in a single layer unlike the papillary ependymomas that have multiple layers. 25% have calcification. There may be nuclear atypia and rare mitoses, but no mucin. There is rarely bone, cartilage, or melanin formation (Figs. 3.77 – 3.79). Fig. 3.77 Choroid plexus papilloma. (A) Sagittal, (B) axial nonenhanced and (C) coronal enhanced T1-weighted magnetic resonance images demonstrating a left lateral ventricular lesion (arrow). (From Albright AL, Pollack IF, Adelson PD, eds. Principles and Practice of Pediatric Neurosurgery. New York, NY: Thieme; 1999. Reprinted by permission.) Fig. 3.78 Choroid plexus papilloma. (A) Axial nonenhanced and enhanced and (B) sagittal enhanced T1-weighted magnetic resonance images demonstrate a fourth ventricular enhancing mass (more common in the left lateral ventricle in children and the fourth ventricle in adults). Fig. 3.79 Choroid plexus papilloma (hematoxylin and eosin [H&E] stain). Fairly well-ordered columnar cells resting on a delicate fibrovascular stroma. d. Immunohistochemistry is positive for transthyretin, vimentin, keratin, S100, and GFAP. They may rarely invade the underlying brain even with benign pathologic findings and may seed the CSF. e. Symptoms are usually from hydrocephalus because of increased CSF production or more likely due to blockage of CSF flow from hemorrhages or direct obstruction. The prognosis does not correlate with the pathologic findings because even benign-appearing tumors may act aggressively. f. Surgical resection usually has a good outcome, although there may be recurrence. There are rare malignant transformations. Both intraventricular meningiomas and choroid plexus papillomas are more frequently on the left. g. The normal choroid is formed from the tela choroidea (the zone of ependymal–pial apposition associated with a fibrovascular stroma) along the choroid fissure at the floors of the lateral ventricles, the roof of the third ventricle, and the lateral recesses of the fourth ventricle. The tela choroidea is composed of vascular tufts covered by choroid epithelium from the ependyma. The choroid plexus may contain benign cysts or xanthogranulomas. h. Choroid plexus carcinoma—accounts for 15% of choroid plexus tumors. They usually occur before 10 years of age, median age is 2 years; they are rare in adults (consider metastasis, especially if they are positive for EMA and negative for S100 and vimentin). Most are located in the lateral ventricles and locally invade the parenchyma, as well as spread throughout CSF pathways. The pathologic findings are less organized, with piled-up epithelium, anaplasia, and necrosis. Treatment is with surgery and radiation with or without chemotherapy. The prognosis is poor (Fig. 3.80). Fig. 3.80 Choroid plexus carcinoma. (A,B) Axial nonenhanced and enhanced T1-weighted magnetic resonance images demonstrating a large enhancing left intraventricular lesion with parenchymal extension. (From Albright AL, Pollack IF, Adelson PD, eds. Principles and Practice of Pediatric Neurosurgery. New York, NY: Thieme; 1999. Reprinted by permission.) C. Mixed neuronal and glial tumors (usually a good prognosis) 1. Ganglioglioma—contains both neoplastic neurons and glial cells. 70% occur before 30 years of age. It is usually in the temporal lobe and most commonly presents with seizures. It is well circumscribed, cystic, firm, and often has a calcified nodule. It may enhance. Pathologic examination demonstrates perivascular inflammatory cells, reticulin, glia, and binucleate neurons with rare mitoses. Immunohistochemistry is positive for neurofilament, synaptophysin, neurosecretory granules, and GFAP (Fig. 3.81 and Fig. 3.82). Fig. 3.81 Ganglioglioma. (A) Axial T1-weighted and (B) coronal enhanced magnetic resonance images demonstrate a right temporal cystic mass with mural nodular enhancement. Fig. 3.82 Ganglioglioma (hematoxylin and eosin [H&E] stain). Clusters of abnormal appearing neurons (some are binucleate, arrow) in a background of neoplastic glial tissue. 2. Gangliocytoma—neoplastic neurons without neoplastic glia. They may be simply dysplastic brain. 3. Desmoplastic infantile ganglioglioma—rare and usually occur before 18 months. They are massive, frontal, cystic lesions adherent to the dura with a desmoplastic reaction. The tumor enhances. It is differentiated from meningioma because it is GFAP positive and EMA negative (Fig. 3.83). Fig. 3.83 Desmoplastic infantile ganglioglioma. (A) Axial T1-weighted and (B) coronal enhanced magnetic resonance images demonstrating a large left-sided posterior frontal-parietal cystic mass with mural nodular enhancement in a child. 4. Dysembryoplastic neuroepithelial tumor (DNET)—usually in people 1–19 years of age, presents with seizures and is located in the temporal lobe. It is circumscribed, cystic, multinodular, superficial, and cortical. It contains normal neurons with abnormal oligodendrocytes and astrocytes (a ganglioglioma has abnormal neurons, is in the white matter, and lacks nodularity). It is associated with cortical dysplasia. Surgical resection is usually curative, and radiation is not needed (Fig. 3.84). 5. Central neurocytoma—occurs in young adults, usually originates at the septum pellucidum, and occurs in the lateral and third ventricles near the foramen of Monro. It is circumscribed, lobulated, enhancing, noninfiltrative, and usually contains calcifications. Pathologic examination demonstrates monotonous hypercellularity similar to oligodendrogliomas with rare mitoses, frequent cysts, and occasionally hemorrhages. Immunohistochemistry is positive for synaptophysin (Fig. 3.85 and Fig. 3.86). Fig. 3.84 Dysembryoplastic neuroepithelial tumor (hematoxylin and eosin [H&E] stain). Oligodendroglioma-like cells with axon bundles surrounding microcystic spaces in which large ganglion cells lie (i.e., “floating neurons”). Fig. 3.85 Central neurocytoma. (A) Axial T1-weighted nonenhanced and (B) enhanced magnetic resonance images demonstrate an enhancing mass near the septum pellucidum and foramen of Monro. Fig. 3.86 Central neurocytoma (hematoxylin and eosin [H&E] stain). Closely packed uniform cells with small blue nuclei and perinuclear halos. The histologic findings are similar to oligodendrogliomas, but central neurocytomas stain with synaptophysin and neuron-specific enolase. D. Primitive neuroectodermal tumors (PNETs) 1. Medulloblast—a term originally coined by Bailey and Cushing in 1925 to describe bipotential cells capable of differentiating into glia or neurons. These cells have features similar to the totipotent neural tube cells. It is postulated that they are derived from the external granular layer of the cerebellum or from dysplastic cell rests in the anterior and posterior medullary velum. The term PNET was introduced by Hart and Earle in 1973. 2. PNET varieties a. Medulloblastoma—50% occur before 10 years and 75% before 15 years, and there is a second peak at 28 years (Fig. 3.87 and Fig. 3.88). There is a male predominance, and they are more frequently off the midline in adults. They account for 20% of CNS tumors in children and one third of the posterior fossa tumors in children. The most common genetic abnormality is isochromosome 17q. It may be associated with Gorlin (basal cell nevus) syndrome that is due to a mutation of the PTCH gene on chromosome 9q. WHO grade IV. Fig. 3.87 Medulloblastoma. (A) Axial computed tomographic nonenhanced and (B) enhanced scans and (C) axial T1-weighted magnetic resonance imaging nonenhanced and (D) enhanced scans demonstrating a slightly hyper-dense and hypointense enhancing mass in the posterior fossa. Fig. 3.88 Medulloblastoma (hematoxylin and eosin [H&E] stain). Closely packed undifferentiated cells with no discernable cytoplasm (small blue cells). b. Retinoblastoma—the most common extracranial malignant solid tumor in children and 80% occur before 5 years. It is derived from a neural crest precursor of the sympathetic ganglia. There is a genetic predisposition by loss of a suppressor gene. It is treated with surgery and radiation. Retinoblastomas contain Flexner-Wintersteiner and Homer Wright rosettes. Trilateral retinoblastoma is bilateral retinoblastoma with pineoblastoma. c. Pineoblastoma (see section I on Pineal Tumors) WHO grade IV d. Ependymoblastoma—WHO grade IV e. Atypical teratoid/rhabdoid tumor—densely cellular blue cell tumors mixed with rhabdoid cells. Rhabdoid cells have eosinophilic rounded, rhabdoid cytoplasmic inclusions. They occur mostly in infants and young children. They are associated with deletions of chromosome 22 containing the INI1/hSNF5 gene; WHO grade IV. f. Central neuroblastoma—usually supratentorial, hemispheric, and circumscribed. It usually occurs before 5 years. It may be hemorrhagic, necrotic, and cystic. The 5-year survival rate is 30%. Neuroblastoma is the third most common tumor in children after leukemia and brain tumor. 2% involve the brain. It is frequently congenital and makes up 18% of tumors in patients <2 months old. Peripheral neuroblastoma develops in the adrenal gland in children and may have spinal epidural metastases. g. Medulloepithelioma—derived from ventricular matrix cells, is the most primitive of the PNETs, and affects very young children; WHO grade IV 3. PNET features a. Locations (in descending order of frequency)—vermis, cerebellar hemispheres (older children), pineal, cerebrum, spinal cord, and brainstem b. PNETs are hyperdense on CT, hypointense on T1-weighted MRI and enhancing (Fig. 3.87). c. They are pink-brown in color and may be soft or firm. They occasionally hemorrhage, are rarely calcified, and are frequently cystic (80%). They have no capsule, are occasionally circumscribed, and frequently disseminate in the CSF. PNETs are densely cellular, contain small round cells with large nuclei and scant cytoplasm, have a variable number of mitoses and occasional necrosis (Fig. 3.88). There are Homer Wright rosettes (around central granulofibrillar material with radially arranged nuclei), pseudorosettes (around blood vessels), ependymal canals (especially with ependymoblastoma), and Flexner–Wintersteiner rosettes (columnar cells with a small lumen seen with retinoblastomas and also pineoblastomas) (Figs. 3.89 – 3.91). There may be a linear array of cells (Indian file) and round islands of cells. There are occasional astrocytes and oligodendrocytes. Neurons can be stained with silver, PTAH, etc. There rarely may be smooth or striated muscle cells or melanocytes. E. Meningiomas—account for 15% of primary intracranial tumors. Peak age is 40–60 years. Females are more commonly affected. 72% of tumors have monosomy 22. 1. Meningioma incidence is increased by radiation and NF2. Fig. 3.89 True “ependymal” rosette (hematoxylin and eosin [H&E] stain). Polar ependymal cells with basal bodies of cilia (blepharoplasts) lining a central lumen; most commonly in ependymoma. (See Fig. 3.29) Fig. 3.90 Homer Wright rosette (hematoxylin and eosin [H&E] stain). Central fibrillar material (processes of tumor cells) ringed by radially arranged cell nuclei; most commonly seen with medulloblastoma and neuroblastoma. Fig. 3.91 Flexner-Wintersteiner rosette (hematoxylin and eosin [H&E] stain). Columnar cells that resemble cone-type photoreceptor cells, which form rosettes with small central lumens; most commonly in pineal tumors and also retinoblastoma. 2. Meningiomas have receptors for progesterone, estrogen, peptides, amines, androgen, glucocorticoids, somatostatin, and cholecystokinin. They may grow with pregnancy and breast cancer. 3. The blood supply is from external carotid artery branches such as the middle meningeal and anterior falcine arteries. They may parasitize pial vessels (from the ICA) and develop a dual supply. They invade dura and bone. Bony changes are hyperostotic more frequently than lytic. The hyperostotic bone usually is invaded by tumor. 4. Meningiomas rarely metastasize. They originate from the arachnoid cap cells (these also form whorls and psammoma bodies) that are most frequently located at the arachnoid granulations (near the superior sagittal sinus under the suture confluence) and the tela choroidea (intraventricular). 5. Locations—cranial (90%), spinal (9%), and ectopic (1%, intraosseous skull, orbit, neck, scalp, sinus, and parotid). Cranial locations in descending order are parasagittal (middle of the superior sagittal sinus), convexity (near the coronal suture), sphenoid ridge, tuberculum, olfactory groove, falcine, foramen magnum, optic nerve, tentorial, choroid (left lateral ventricle), and thoracic. The parasagittal and convexity groups account for 50% (Figs. 3.92 – 3.97). Fig. 3.92 Parasagittal meningioma. (A) Nonenhanced and (B) enhanced coronal and (C) nonenhanced sagittal T1-weighted magnetic resonance imaging scans demonstrating a homogeneously enhancing circumscribed isoin-tense lesion arising from the medial dura and invading the wall of the superior sagittal sinus. Fig. 3.93 Convexity meningioma. (A) Axial enhanced and (B) bone window computed tomographic scans demonstrating a homogeneously enhancing, circumscribed, calcified lesion arising from the convexity. Fig. 3.94 Sphenoid wing meningioma. (A) Sagittal nonenhanced and (B) enhanced T1-weighted magnetic resonance imaging scans and (C) axial enhanced computed tomographic scan demonstrating a homogeneously enhancing circumscribed lesion arising from the sphenoid ridge. Fig. 3.95 Cerebellopontine angle meningioma. (A) Axial nonenhanced and (B) enhanced computed tomo-graphic scans demonstrating the dural-based enhancing left-sided mass. Fig. 3.96 Olfactory groove meningioma. (A) Enhanced sagittal and (B) axial T1-weighted magnetic resonance images demonstrating a homogeneously enhancing circumscribed lesion arising from the floor of the anterior fossa. Fig. 3.97 Tentorial meningioma. Angiogram (lateral view) demonstrates the tumor blush of the meningioma with supply by an enlarged tentorial artery of Bernasconi-Cassinari. (From Huber P, Krayenbühl H, Yasargil MG. Cerebral Angiography. 2nd ed. New York, NY: Thieme; 1982. Reprinted by permission.) 6. They are multiple in 9% of cases. Meta-static neoplasms have been reported inside of meningiomas.
I. General Neuropathology
II. Glial Cell Response to Injury
III. General Neuroradiology
IV. Nervous System Development
V. Developmental Pathology
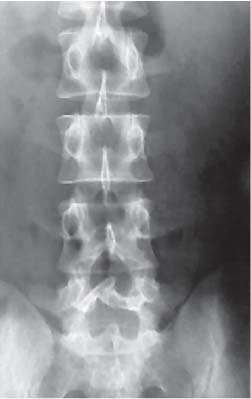
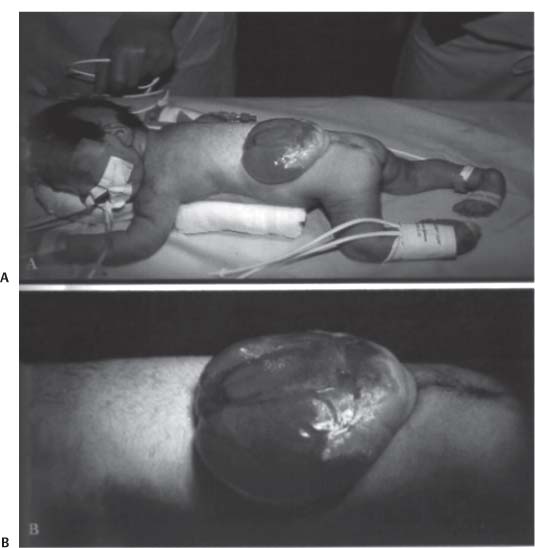
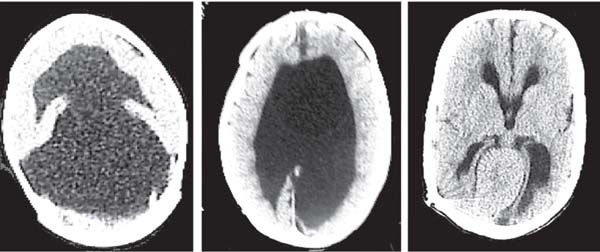
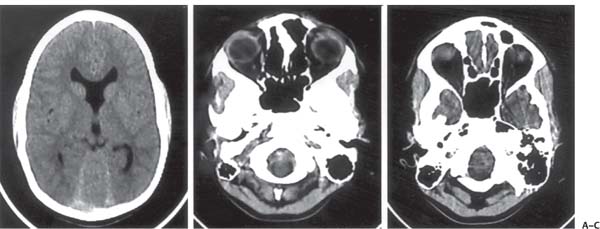
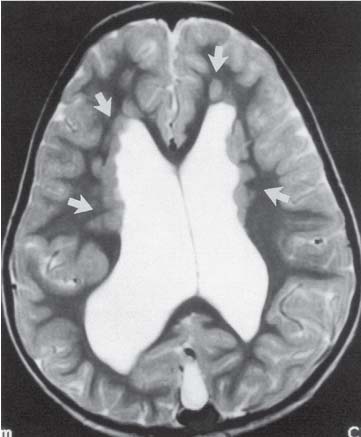
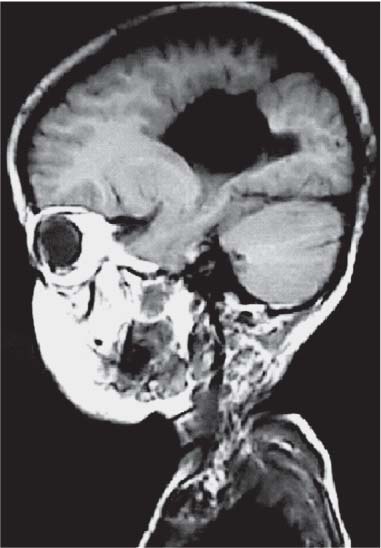
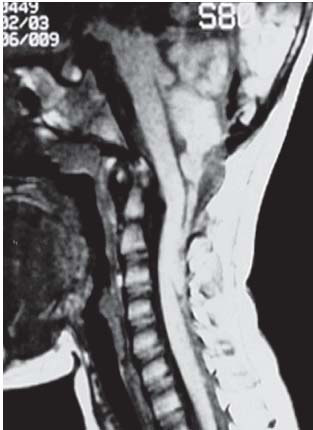
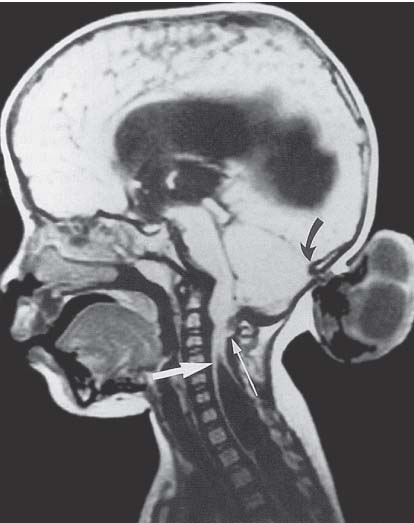
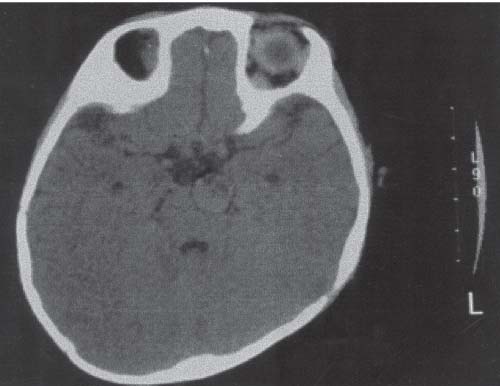
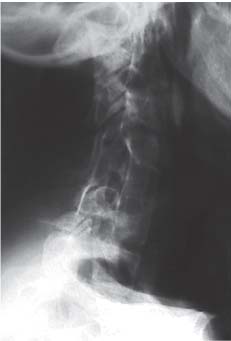
VI. Perinatal Brain Injuries
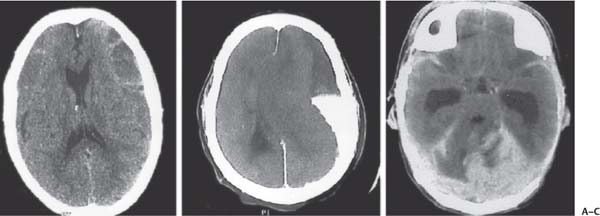
VII. Infectious Diseases
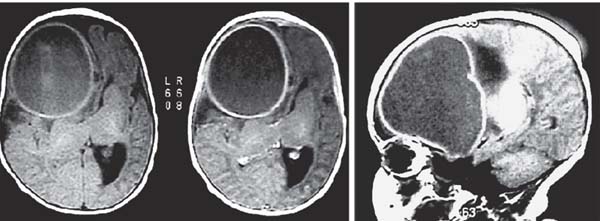

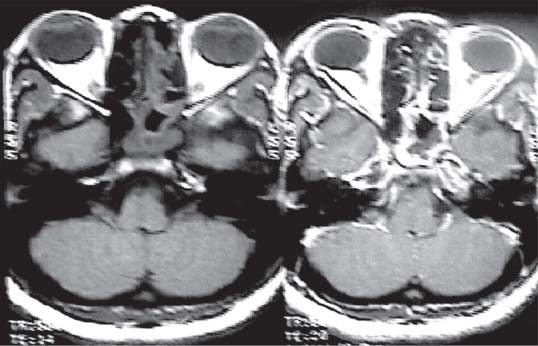
VIII. Infectious Pathogens
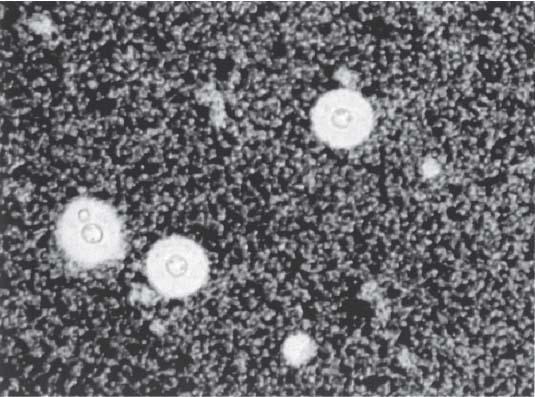
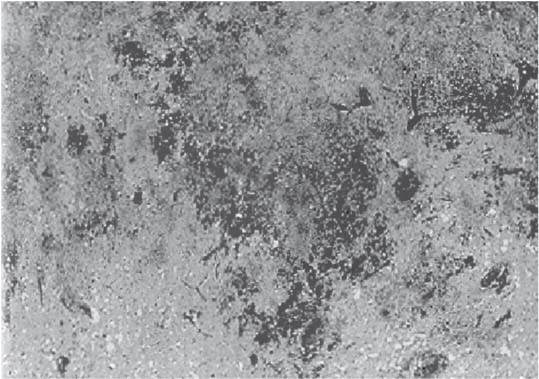
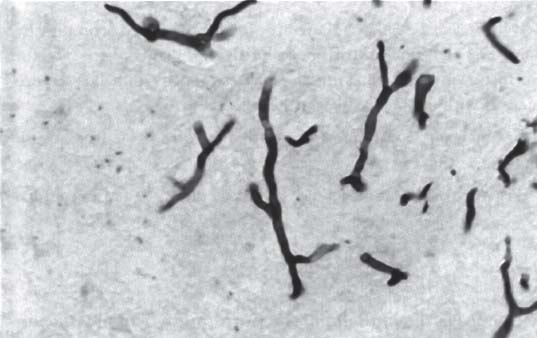
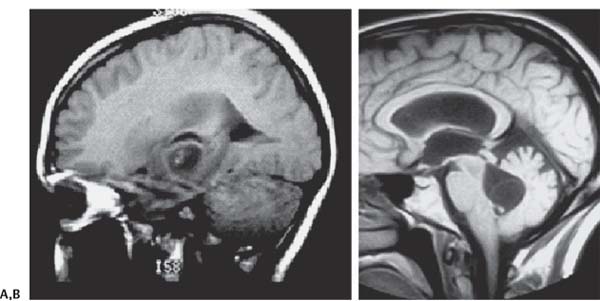
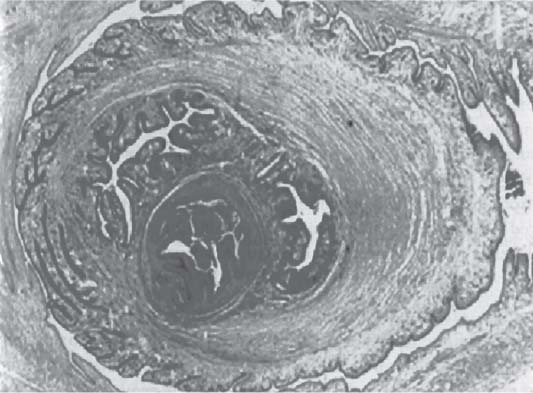
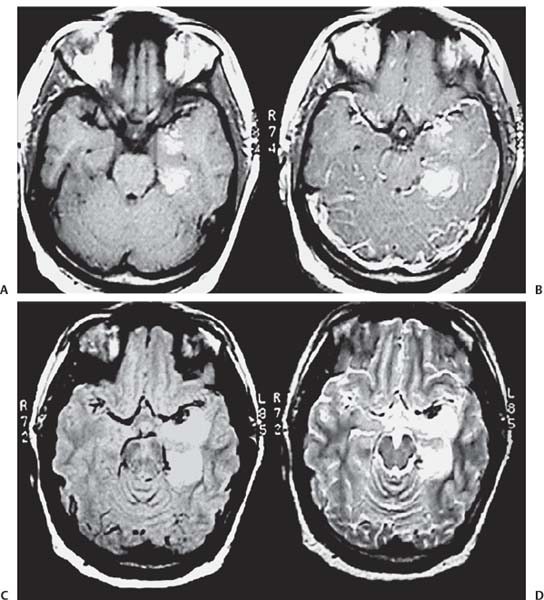
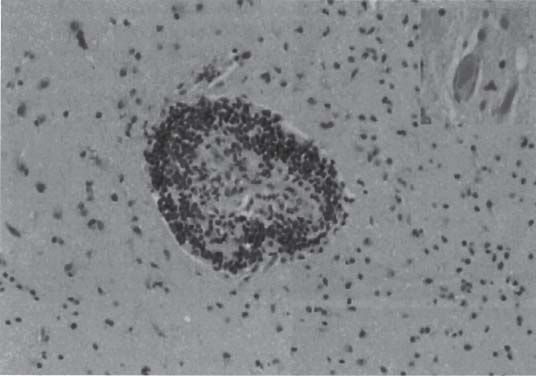

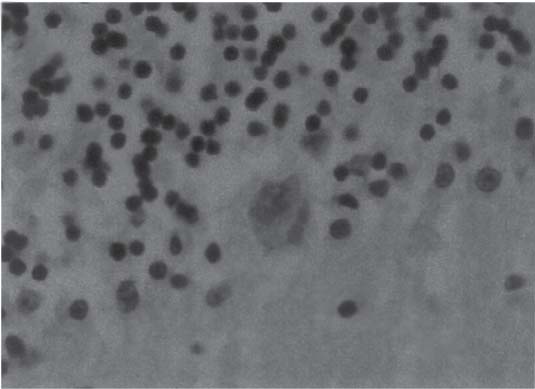
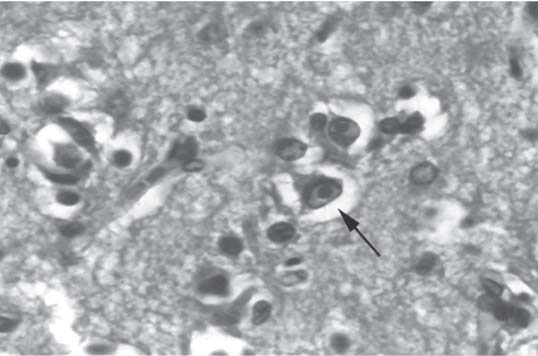
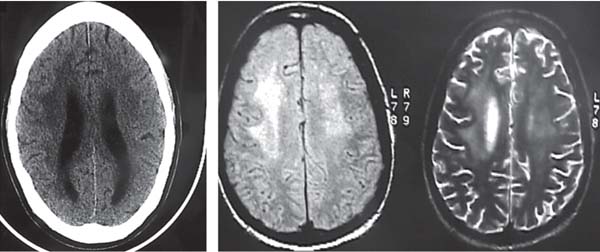
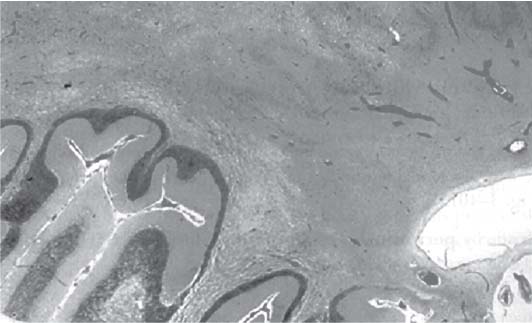
IX. Congenital Infections (Torch)
X. Oncology
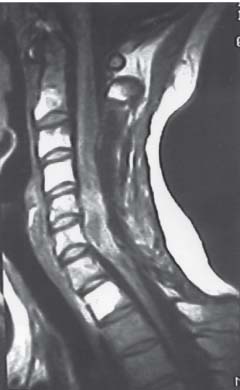
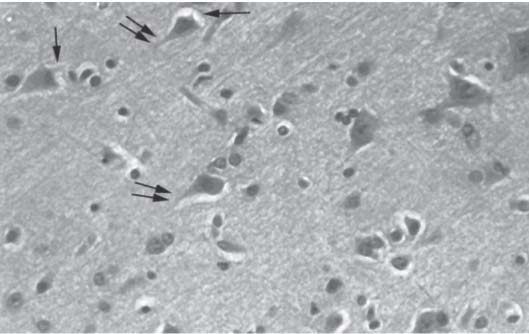
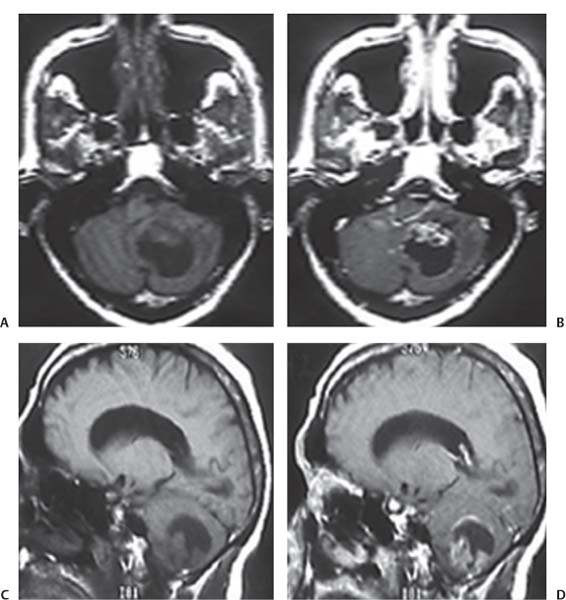
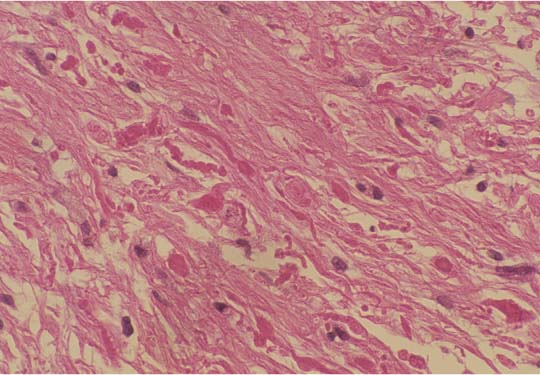
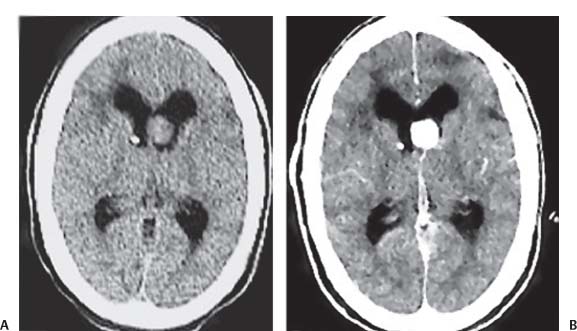
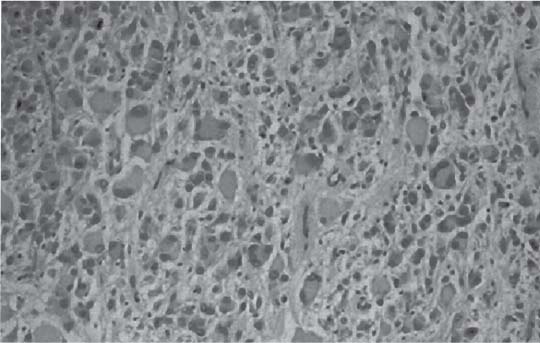
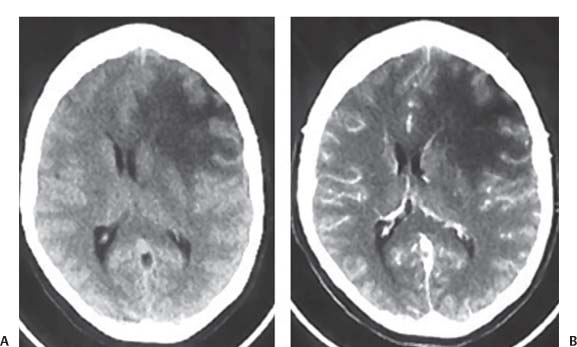
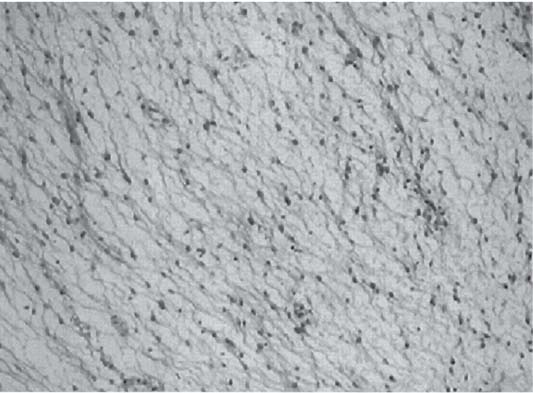
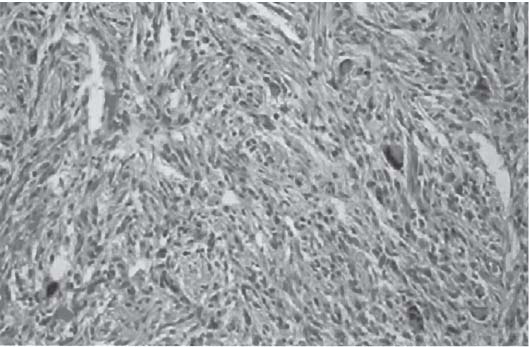
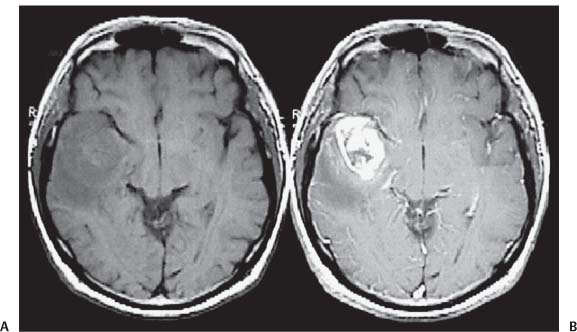
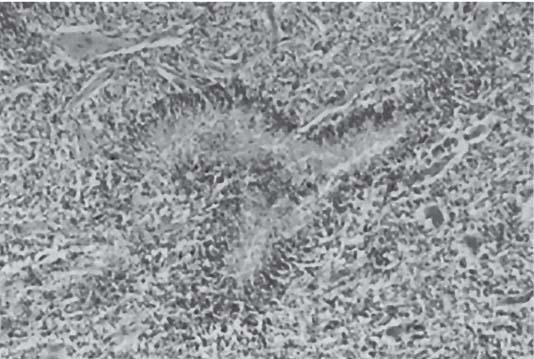
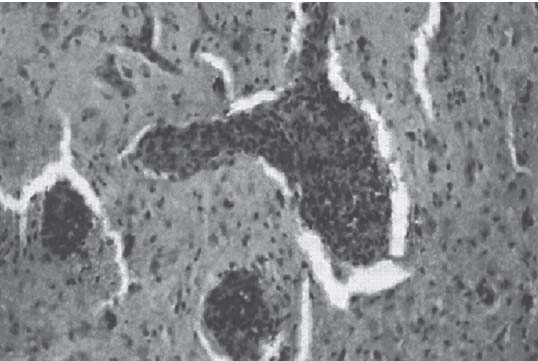

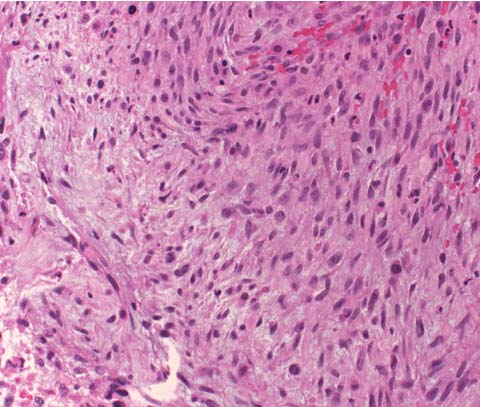
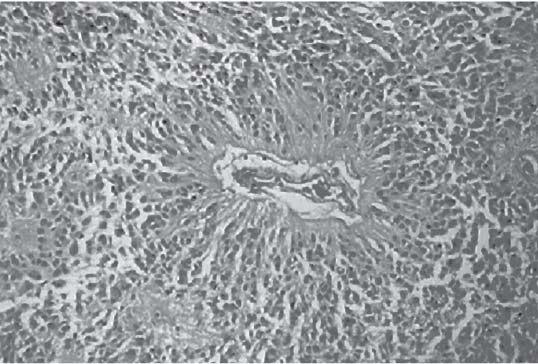
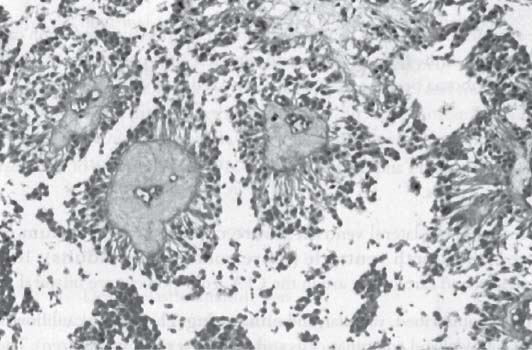
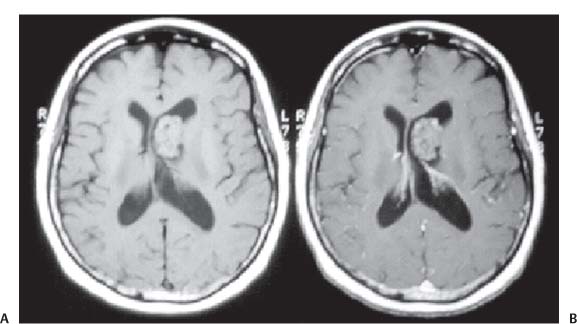
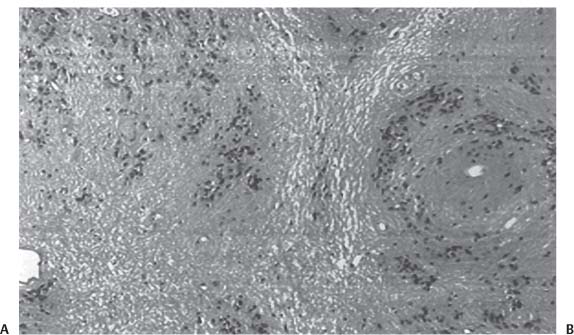
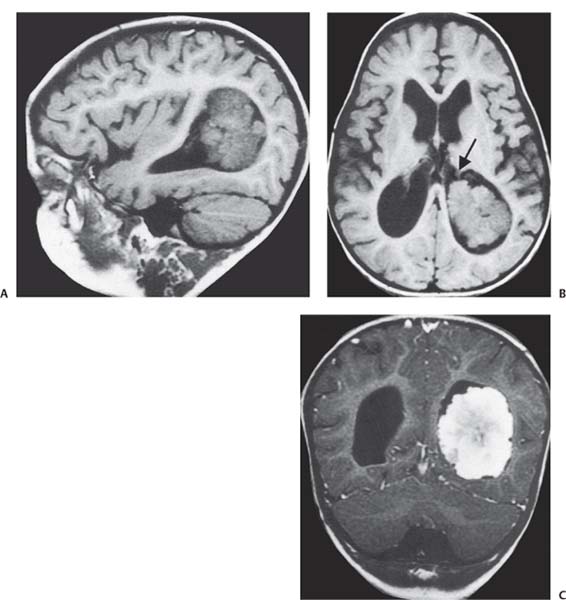
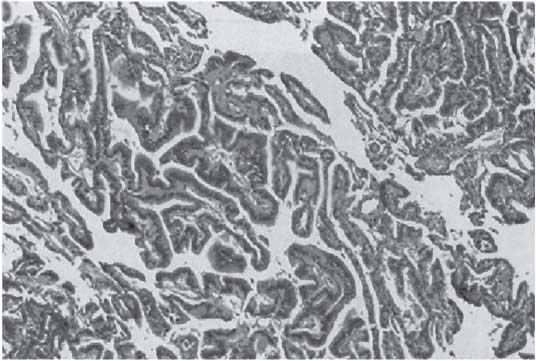
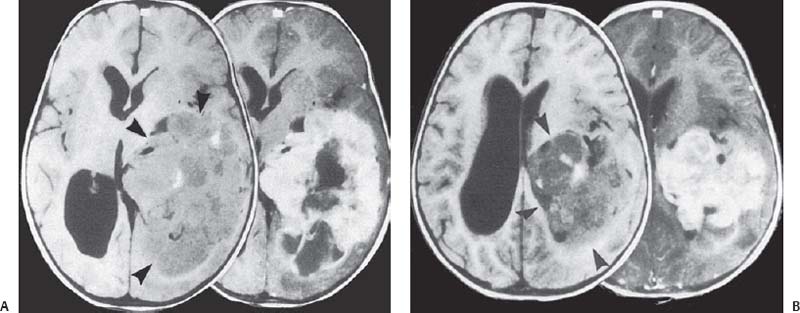
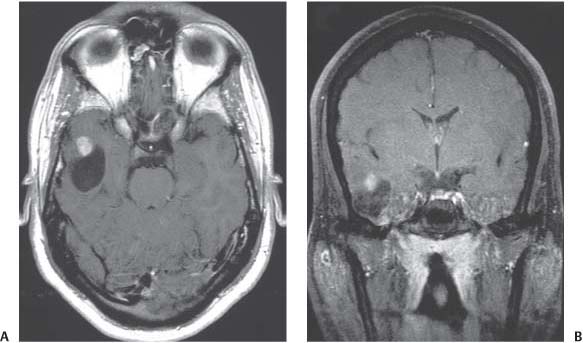
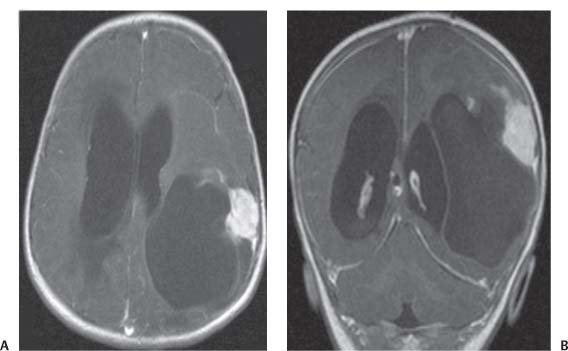
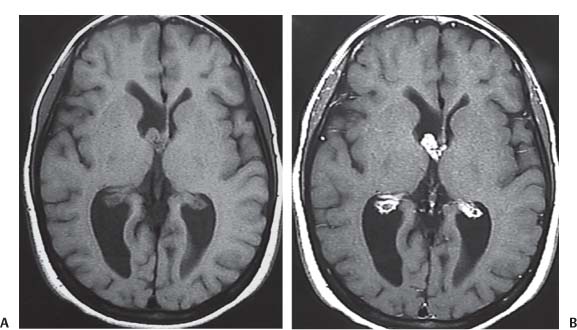
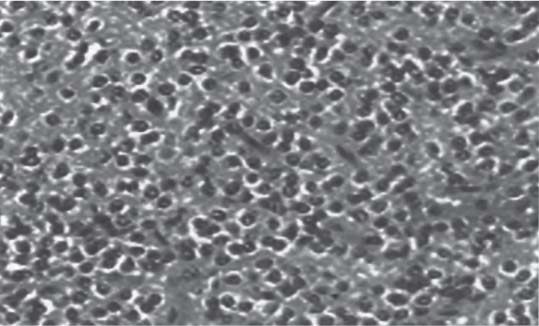
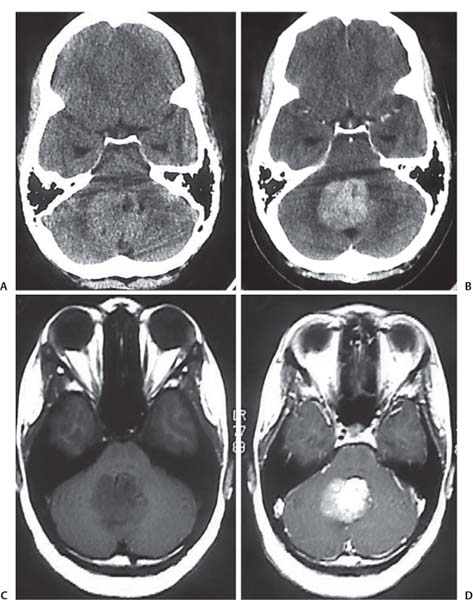
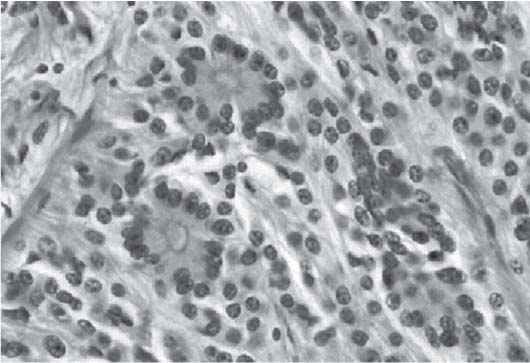
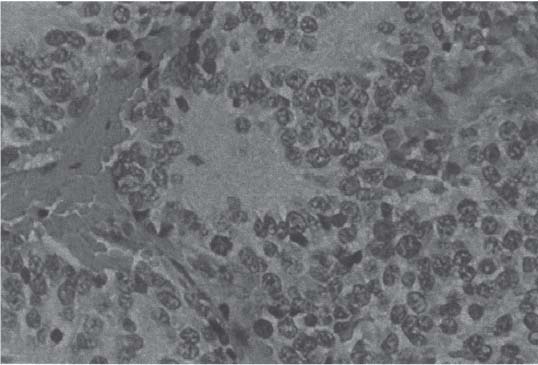
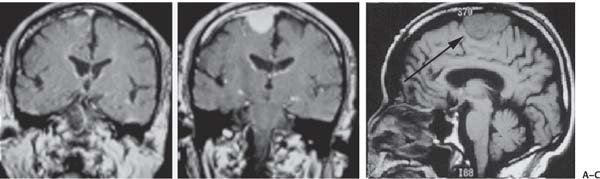
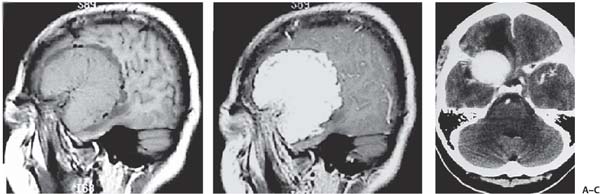
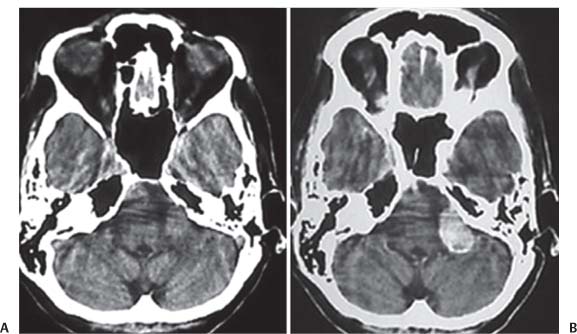
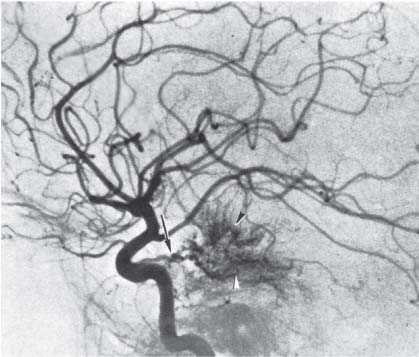
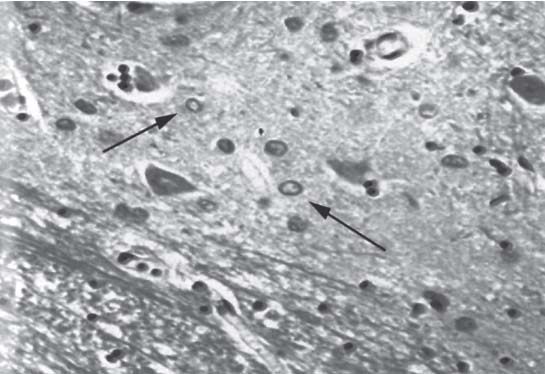
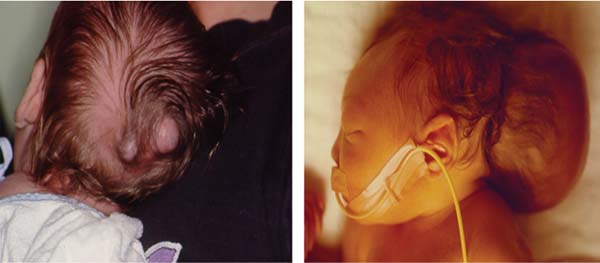
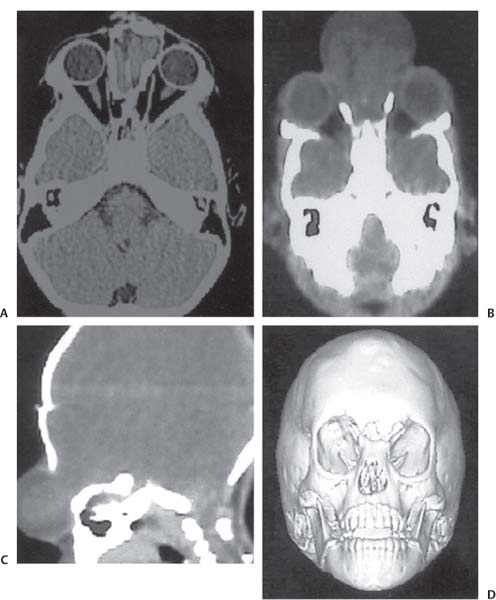
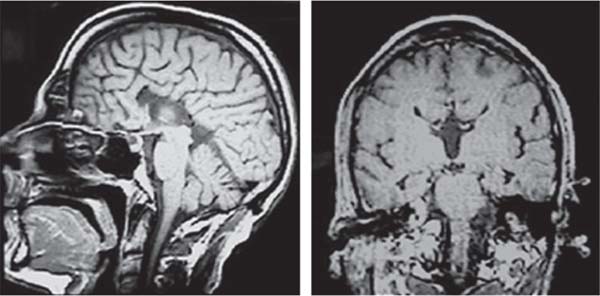
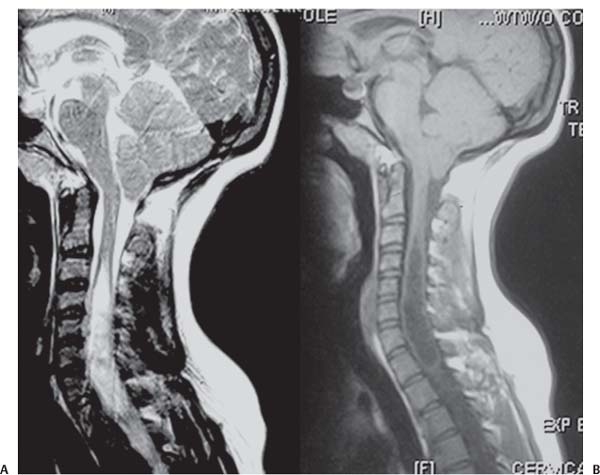
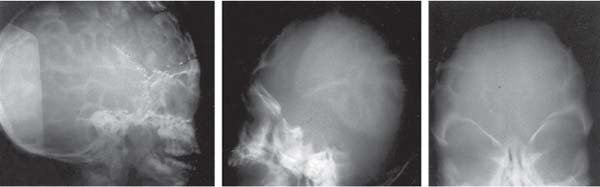
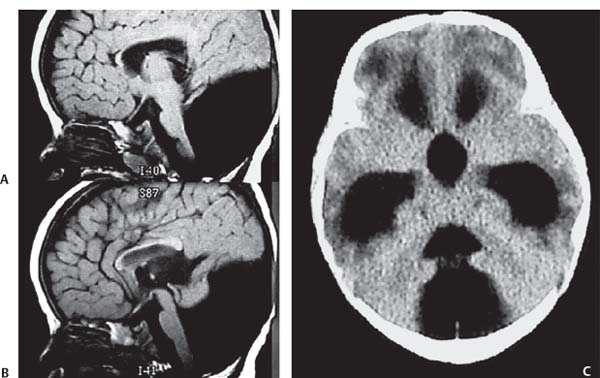
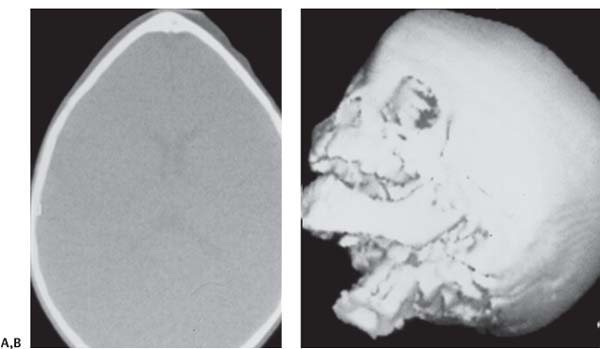
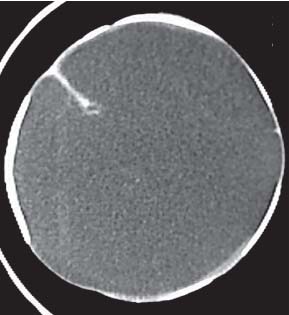
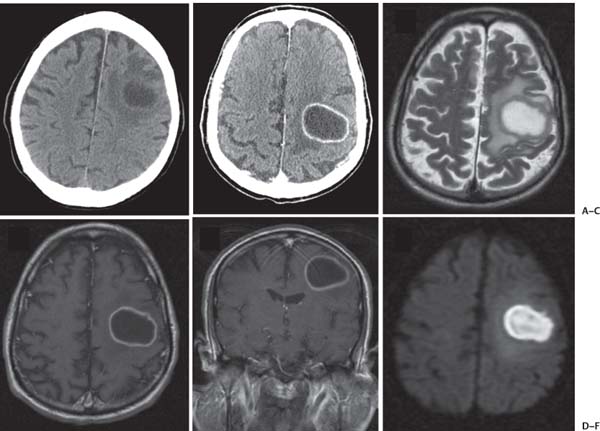
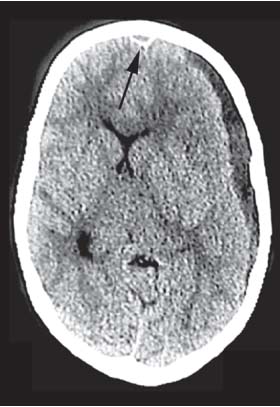

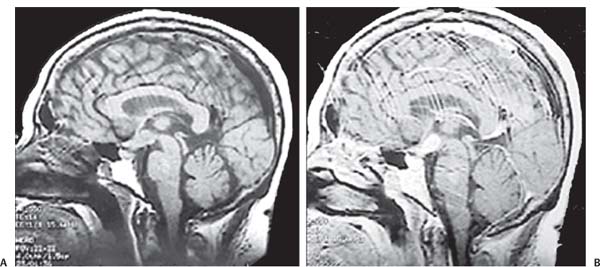
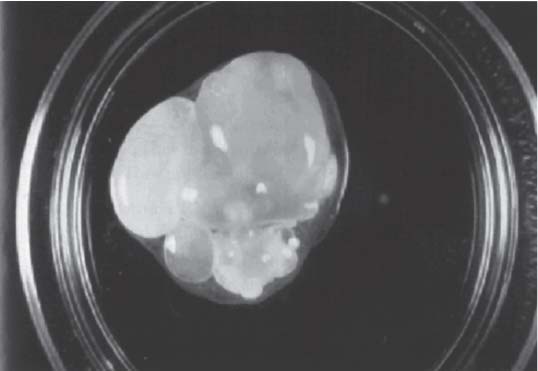
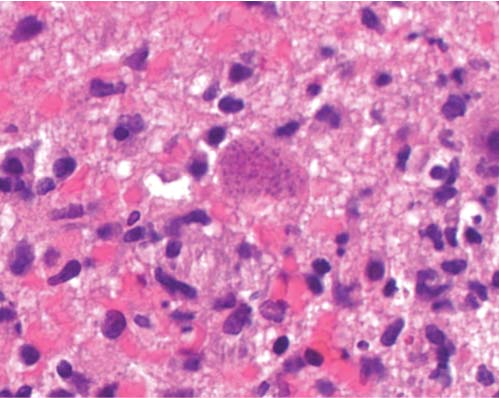
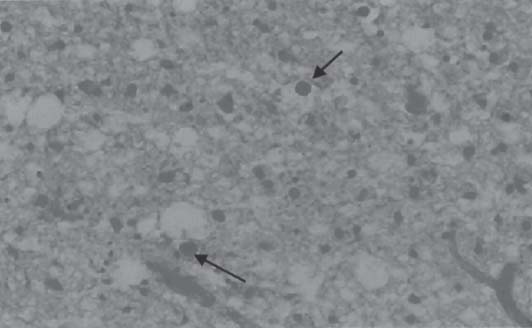
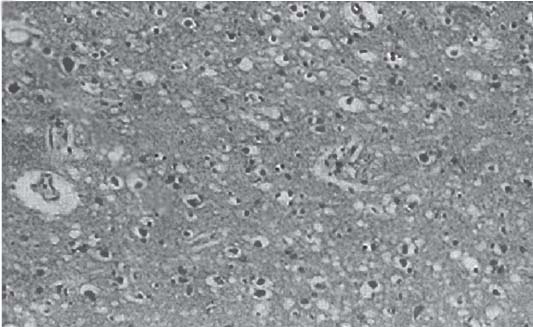
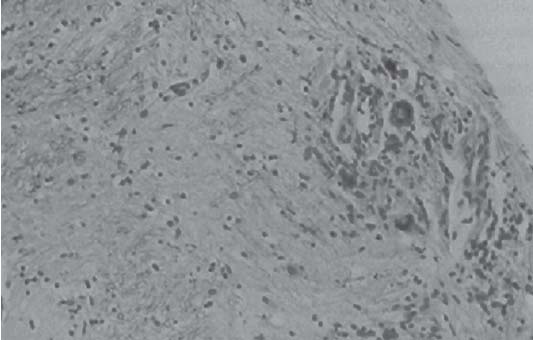
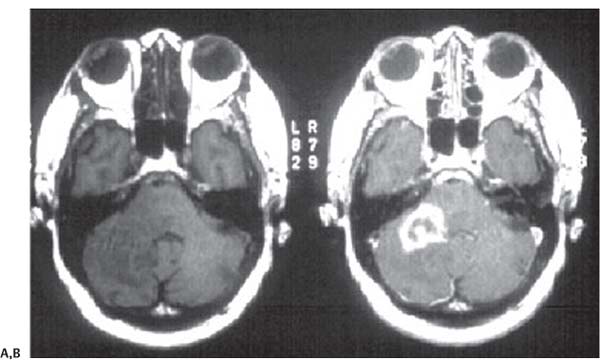
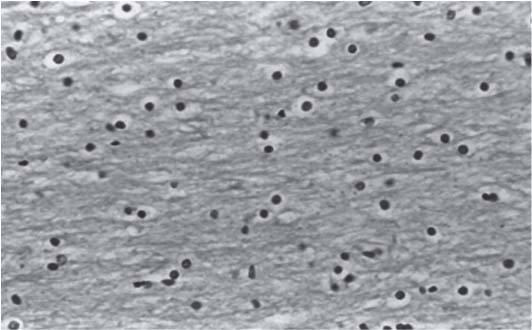
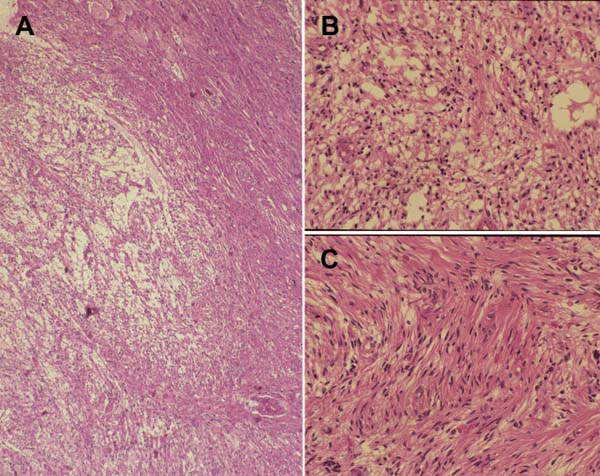
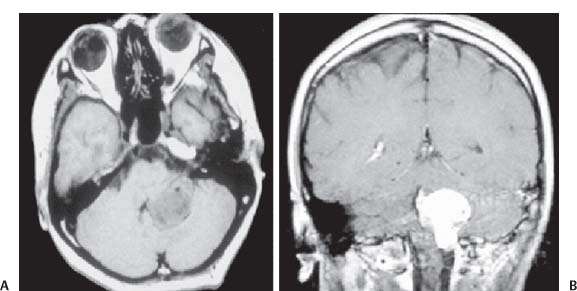
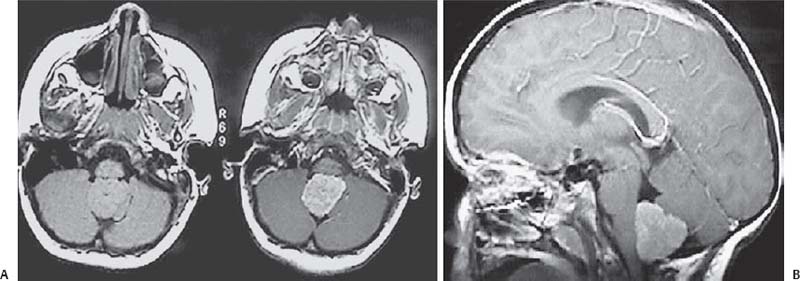
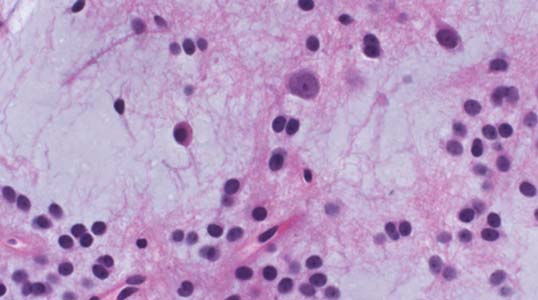
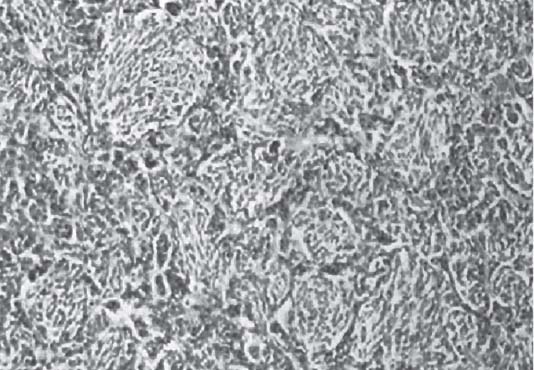
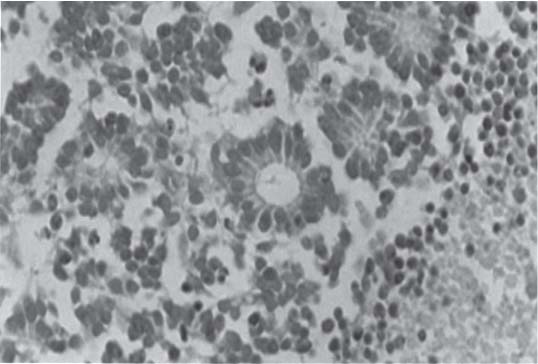
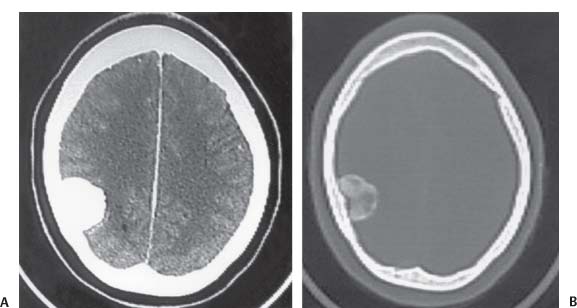
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


