Fig. 25.1
The N-methyl-d-aspartate (NMDA) receptors are heterotetramic complexes composed of four subunits, typically two NR1 subunits (with eight variants by alternative splicing of the GRIN1 gene) and two NR2 subunits (with four splicing variants) as depicted. The incorporation of NMDA receptor (NR) subunit, NR3, into conventional NMDA receptors forms glutamate-activated NR1/NR2/NR3 triheteromers, whereas the omission of the glutamate-binding NR2 subunits results in excitatory glycine-activated NR1/NR3 diheteromers. Combinations of different NR2 subunits determine the pharmacological and physiological profile of the receptor in terms of channel kinetics, and its affinity for agonists and antagonists for conventional NR1/NR2 NMDA receptors. l-glutamate binds to NR2 subunits, whereas glycine binds to the co-agonist, glycine-B site, of the two NR1 subunits. All four sites have to be activated to permit activation of the ion channel that permits the inward flow of Ca2+, Na+ and K+ ions
Glycine as a Modulator of NMDA Receptor
The amino acid, glycine, is an important signalling molecule in the central nervous system (CNS). As an inhibitory neurotransmitter, glycine binds to the strychnine-sensitive glycine-A site on ionotropic glycine receptors and activates an inward chloride current through the integral anion channel of the glycine receptor complex which leads to the hyperpolarization of the postsynaptic membrane. This fast neuronal inhibition is the predominant function of glycine receptor found predominantly at inhibitory synapses in the spinal cord, brainstem, and retina in the adult brain. The existence of non-strychnine-sensitive high affinity binding sites was revealed by [3H] glycine autoradiography because such sites were not labeled by [3H] strychnine [20]. The distribution pattern of the non-strychnine-sensitive, glycine-B sites, corresponds well to the binding profile of NMDA receptors [21]. The physiological interaction between glycine and NMDA receptors was demonstrated by Johnson and Ascher [22]. They showed that glycine could augment NMDA receptor-mediated electrophysiological responses. Kleckner and Dingledine [23] subsequently demonstrated that glycine is a prerequisite for the activation of the NMDA receptor and coined the term “co-agonist” to describe its essential role at the NMDA receptor. It is known that glycine is not the only endogenous ligand at the glycine-B site. The amino acid d-serine can also bind to the glycine-B site and serves the function of an obligatory co-agonist [24–26]. Hence, the concomitant binding of glycine or d-serine at the glycine-B site is a prerequisite for the activation of postsynaptic NMDA receptors by glutamate released from presynaptic terminals. The distinction and cooperation between the regulatory function of d-serine and glycine at NMDA receptors, however, remain to be fully delineated. One suggestion is that d-serine might be preferentially involved in the regulation of synaptic NMDA receptors, whereas glycine appears to be more critically involved in the regulation of extra-synaptic NMDA receptor excitability—a demarcation that can be linked to the differential distributions of synaptic versus extra-synaptic NMDA receptors, which can be distinguished by the presence of NMDA receptor (NR) subunit NR2A and NR2B, respectively.
Occupancy of the glycine-B site is not only required for initiating signaling through the NMDA receptor, but it also primes the NMDA receptor for internalization [27]. Saturation of the glycine-B sites is expected to trigger the internalization of NMDA receptors, and thereby would be expected to weaken the signals mediated by NMDA receptors. This could take place when the extra-cellular glycine concentration is sufficiently high. It has been shown that the magnitude of NMDA receptor-dependent long-term potentiation can be enhanced by increasing concentration of ambient glycine up to 10 μM, but signs of impairment can appear at ≥100 μM concentrations [28]. Therefore, disturbance of the homeostatic regulation of extracellular glycine concentration in the brain could have a profound effect not only on the excitability of individual NMDA receptors but also on their overall expression levels.
Modulation of NMDA Receptor Through Glycine Transporters
The ambient concentration of glycine in the extracellular space near NMDA receptors may be exploited as a new avenue to facilitate NMDA receptor neurotransmission [29]. One obvious means to increase extracellular glycine availability is the direct augmentation through dietary intake of glycine. However, this proves to be ineffective because of the poor penetration of glycine through the blood–brain-barrier (see section “Proof of principle I”). Alternatives include glycine-B site partial agonist, d-cycloserine, which is able to penetrate into the CNS. A potential breakthrough hinges on the homeostasis (re-uptake, release, and metabolism) of glycine in the brain.
It was initially believed that the levels of glycine in the extracellular space were sufficiently high so that the glycine-B sites of the NMDA receptors would be saturated under physiological conditions [30, 31] and therefore not relevant in the normal regulation of NMDA receptor function. It soon became apparent that glycine transporters provide active removal of extracellular glycine such that the glycine concentration in the synaptic cleft is maintained at sub-saturating levels [32–34].
The two principal subtypes of glycine transporters are GlyT1 [35, 36] and GlyT2 [37]. They belong to the sodium-dependent intracelluar solute carrier family 6 of transporters but differ in terms of 1) regional and cellular expression patterns in the CNS, 2) Na+:glycine stoichiometries, and 3) the ability to reverse-transport intracelluar glycine into the extracellular space [38–42]. Five variants of GlyT1 (a–e) and three variants of GlyT2 (a–c) as a result of alternative splicing and promotor usage have been identified [40, 43–45].
As illustrated in Fig. 25.2, GlyT2 expressed in glycinergic terminals assumes the critical role in the reuptake and recycling of glycine released by the pre-synaptic terminal into the synaptic cleft, and together with astrocytic GlyT1 contribute to the termination of the stimulation of glycinergic receptors in the post-synaptic membrane [40]. The coordinated activities of GlyT1 and GlyT2 are essential for the vital functions that depend on inhibitory neurotransmission such as respiratory regulation. The loss of pre-synaptic GlyT2 drastically curtails the refilling of glycine vesicles by the presynaptic cell and severely undermines inhibitory glycinergic neurotransmission [42, 46, 47]. On the other hand, the loss of astrocytic GlyT1 severely undermines the clearance of glycine released from the synaptic cleft leading to sustained neuronal inhibition over brain stem respiratory centers [48]. In fact, constitutive homozygous deletion of either the GLYT1 or GLYT2 gene is lethal in mice [46, 48].
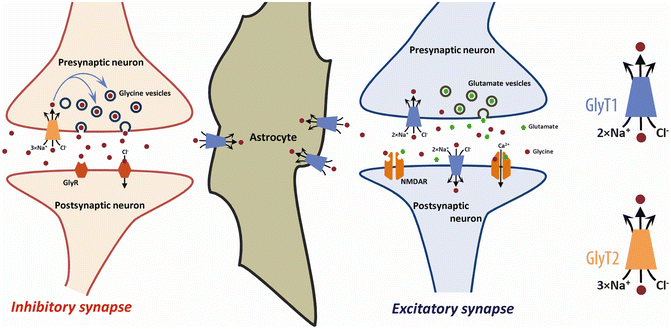
Fig. 25.2
Regulation of glycinergic neurotransmission at inhibitory and excitatory synapses by glycine transporters. At an inhibitory synapse (left) containing glycine receptors, GlyRs, the extracellular level of glycine is regulated by both GlyT1 and GlyT2. The activation of postsynaptic GlyRs by glycine released from the presynaptic bouton is terminated by the re-uptake of glycine by GlyT2 expressed in the presynaptic terminals and GlyT1 in adjoining astrocytes. Glycine transported back in the pre-synaptic terminals by GlyT2 is incorporated into presynaptic vesicles by the vesicular inhibitory amino acid transporter (not shown). At the excitatory synapse (right), GlyT1 is located on pre- and post-synaptic neurons, where it co-localizes with N-methyl-d-aspartate (NMDA) receptors, as well as on neighbouring astrocytes. GlyT1 regulates the extracellular glycine concentration at the synaptic cleft and keeps the glycine levels below what is required to saturate the glycine-B site. Inhibition of GlyT1-mediated glycine reuptake near glutamatergic synapses is highly effective in elevating the baseline occupancy of the glycine-B site and thereby increases the probability of NMDA receptor responses
In contrast, GlyT1 expressed in pre- and post-synaptic sites of glutamatergic synapses plays the pivotal role in preventing the saturation of the glycine-B site on NMDA receptors, with GlyT1 expressed in adjoining astrocytes as well as extrasynaptic sites playing a supplementary role [33, 34, 49–52]. GlyT1 is therefore the obvious target of choice to modify NMDA receptor function. Inhibition of GlyT1 should elevate baseline glycine-B site occupancy and thereby increase the probability of NMDA receptor activation by presynaptic release of glutamate (Fig. 25.1). The result is an ‘on-demand’ facilitation of NMDA receptor activation which amplifies the glutamatergic signals. This is unlike the elevation of the tonic activity of NMDA receptors produced by direct NMDA receptor agonists. GlyT1 inhibition is therefore expected to confer therapeutic potential for diseases such as schizophrenia, in which deficient signaling via forebrain NMDA receptors is implicated. We have shown that the deletion of neuronal GlyT1 in the forebrain alone is sufficient to enhance selectively the NMDA receptor current; and the knockout mice have exhibited antipsychotic-like as well as pro-cognitive phenotypes [53, 54].
Schizophrenia could be the first neuropsychiatric disorder to benefit from this novel pharmacological approach. The recent phase II clinical trials of the GlyT1 inhibiting drug, bitopertin, [4-(3-Fluoro-5-trifluoromethylpyridin-2-yl)piperazin-1-yl]-[5-methanesulfonyl-2-((S)-2,2,2-trifluoro-1-methylethoxy)phenyl]methanone] (a.k.a RO4917838 and RG1678) developed by Hoffmann-La Roche and its subsidiary Chugai Pharmaceutical have yielded promising outcomes for the treatment of schizophrenia negative and cognitive symptoms [55–58]. The compound had the potential to become the first successful translation of the glutamate hypofunction hypothesis of schizophrenia into a clinic-ready pharmacotherapy since the hypothesis’s inception in the 1980s [59] before the hope was dashed finally with the release of the disappointing results. First, two trials were reported to have failed to meet the primary endpoint in improving negative symptoms (www.roche.com/med-cor-2014-01-21-e.pdf). Subsequently, more information were released in abstract form in three international conferences [60–62]. Out of the six phase III trials, only one treatment arm (10 mg/kg adjuvant biotopertin) in the study, which had targeted positive symptoms, had yielded significant improvement the primary endpoint relative to placebo adjuvant treatment. The evidence was too weak to justify further commercial development of bitopertin, and Roche has since shelved its development as an anti-schizophrenia agent, although it is still being pursued as an adjuvant treatment for obsessive compulsive disorder (NCT01674361).
Pharmacological Models
The development of potent and highly selective GlyT1 inhibitors has been initiated with the synthesis of the sarcosine derivatives, Org 24598 and N[3-(4’-fluorophenyl)-3-(4’-phenylphenoxy)propyl]sarcosine (NFPS; also known as ALX 5407 since 2001) [63, 64]. A variety of compounds have since been synthesized, including compounds that are not based on a sarcosine backbone such as SSR504734, SSR103800, and bitopertin [53, 65]. As proof of mechanism, GlyT1 inhibitors have been demonstrated to elevate brain glycine levels, potentiate NMDA receptor-mediated transmission and synaptic plasticity, and antagonize the behavioral effects of NMDA receptor blockade [53].
The first preclinical evidence of antipsychotic-like potential of glycine re-uptake inhibition can be traced back to the study by Toth et al. [66], which showed that the glycine derivative, glycyldodecylamide, was much more effective than glycine itself to reverse the hyperlocomotor response induced by the NMDA receptor antagonist, phencyclidine. However, it took over a decade until the connection with glycine transporter was identified by Javitt and colleagues. They demonstrated that behaviorally effective doses of glycyldodecylamide were highly effective in inhibiting forebrain glycine re-uptake [67, 68]. That early finding marked the beginning of the preclinical evaluation of many GlyT1 inhibitors in animal models of schizophrenia ranging from the readily available compound sarcosine to highly potent synthetic compounds developed by diverse pharmaceutical companies. As summarized in Table 25.1, the results obtained in preclinical animal models have been largely encouraging.
Table 25.1
Overview of major psychopharmacological findings of selective GlyT1 inhibitors in pre-clinical schizophrenia models
Compound | Locomotor activity | Prepulse inhibition (PPI) | Latent inhibition (LI) | Effects on memory | Others |
|---|---|---|---|---|---|
Manufacturer | |||||
ALX 5407 (a.k.a. NFPS) | • Attenuates PCP-induced hyperactivity • Fails to attenuate amphetamine-induced hyperactivity • Fails to attenuate apomorphine-induced hyperactivity | • Enhances PPI in DBA/2 mice • Attenuates PPI in C57BL/6 mice • Reverses MK801-induced PPI disruption | • Enhances LI expression • Normalizes MK801-induced LI abnormalities (namely, both disruption and persistent LI) | • Ameliorates MK801-induced spatial reference memory but not working memory • Enhances social recognition memory • Reverses MK801/PCP-induced memory deficits • Facilitates extinction learning of conditioned fear | |
ASP2535 Astellas | • Reverses PCP-induced PPI disruption | • Reverses MK-801- and scopolamine-induced working memory deficit • Restores recognition memory impairment induced by neonatal PCP treatment | |||
Merck (S) Merck | • Enhances PPI in DBA/2 mice | ||||
ORG 24461 Organon | • Attenuates PCP- and amphetamine-induced hyperactivity • Fails to attenuate apomorphine-induced stereotypy | ||||
ORG 24598 Organon | • Restores PPI deficit in the rat model of neonatal ventral hippocampal lesions | ||||
PF-3463275 Pfizer | • Reverses ketamine-induced memory deficits | ||||
RG1678 Hoffman-La Roche | • Attenuates PCP-induced hyperactivity • Attenuates amphetamine-induced hyperactivity | ||||
Roche-7 Hoffman-La Roche | • Enhances PPI in DBA/2 mice | ||||
SSR103800 Sanofi | • Attenuates MK801-induced hyperactivity | • Enhances PPI in DBA/2 mice | • Enhances LI expression • Normalizes amphetamine- and MK801-induced LI abnormalities | • Reverses social memory deficit induced by chronic neonatal PCP exposure • Reverses object memory deficit in PCP-sensitized rats | • Antidepressant-like effect in the forced swim test |
SSR504734 Sanofi | • Attenuates PCP- and MK801-induced hyperactivity • Exacerbates amphetamine -induced hyperactivity • Potentiate the hypoactivity effect induced by low doses of apomorphine • Reverses hypersensitivity to amphetamine induced by neonatal chronic PCP exposure | • Enhances PPI in DBA/2 or C57BL/6 mice • Exacerbates the disruption of PPI induced by apomorphine | • Enhances LI expression • Normalizes amphetamine- and MK801-induced LI abnormalities | • Reverses social memory deficit induced by chronic neonatal PCP exposure • Enhances working memory performance in a continuous delayed alternation task | • Suppresses acquisition of contextual fear (anxiolytic effect?) • Facilitates extra-dimensional shift (improved cognitive flexibility?) |
Reversal of the hyperlocomotor activity induced by NMDA receptor blockers such as phencyclidine and dizocilpine (MK-801) is most commonly used as antipsychotic potential test (Table 25.1). In an effort to more directly measure the in vivo efficacy of a given compound to enhance the occupancy of the NMDA receptor glycine-B sites, Alberati et al. [69] instead measured a given compound’s ability to reverse the motor disturbance (head weaving, body rolling, hyperlocomotion) induced by l-687,414. As a partial agonist, l-687,414 is less efficacious than the endogenous agonists, glycine or d-serine, at the co-agonist glycine-B site; and therefore l-687,414 can induce behavioral effects resembling direct NMDA receptor antagonists. Hence, a drug that can effectively raise the levels of the endogenous co-agonist, glycine, could reverse the behavioral effects of l-687,414 because the increase in glycine directly competes with l-687,414 for binding to the glycine-B site. The test is uniquely suited for the detection of potential antipsychotic drugs that act to increase endogenous glycine-B site occupancy. At doses that were devoid of notable behavioral effects, glycine and five potent GlyT1 inhibitors (ALX 5407, ORG24598, RO4543338, RO4840700, and SSR504734) dose-dependently reversed l-687,414-induced hyperlocomotion. Additionally, the assay is sensitive to conventional antipsychotics, but not to other CNS drugs such as the analgesic morphine, the antidepressant fluoxetine, or the anxiolytic drug diazepam [69].
It is worth noting that the ability of GlyT1 inhibitors to antagonize the motor stimulant effect of the indirect dopamine agonist, amphetamine—a model of the positive symptoms associated with hyperdopaminergia [70, 71], is less consistent than their ability to reverse the hyperlocomotor effects effect of NMDA receptor blockade (see Table 25.2). One competitive GlyT1 inhibitor, SSR504734, has even been reported to exacerbate the hyperlocomotor response of amphetamine [72] and the sensorimotor gating deficits in the prepulse inhibition paradigm induced by apomorphine (a direct dopamine receptor) [73]. These drug–drug interactions are consistent with a report that SSR50473 potentiated the release of dopamine in the nucleus accumbens triggered by direct electrical stimulation of the amygdala [74] and increased basal dopamine activity in the prefrontal cortices [75].
Table 25.2
Overview on clinical trials evaluating the therapeutic efficacy of GlyT1 inhibitors for the treatment of schizophrenia
Compound | Study design and scope | Trial codes | |
|---|---|---|---|
Manufacturer/institutions | |||
Phase 1 | GSK1018921 GlaxoSmithKline | Repeat-dose study to assess safety, tolerability, pharmacokinetics, pharmacodynamics of GSK1018921 in healthy volunteers and patients with schizophrenia | NCT00929370 Terminated 2009 |
PF-3463275 Pfizer | Evaluated the safety, tolerability, and efficacy of two-dose regimens of PF-3463275 compared with placebo added to ongoing atypical antipsychotic therapy for cognitive deficits in subjects with chronic symptoms of schizophrenia | NCT00567203 Completed 2008 | |
PF-04958242 Pfizer | Evaluated the safety and tolerability of multiple, ascending doses of PF-04958242 administered orally to psychiatrically stable subjects with schizophrenia receiving antipsychotic and adjunctive medication | NCT01518894 Completed 2012 | |
SSR504734 and SSR103800 Sanofi | No detailed information has been disclosed on the design or outcome of the Phase I study | Not available | |
Org 25935 Organon | Investigated, in collaboration with Yale University, the effect of Org 25935 on ketamine-induced behavioural and cognitive effects in healthy male subjects. Early results suggested that Org 25935 might exacerbate some cognitive deficits induced by ketamine | NCT00700076 Completed 2008 | |
RO4917838 (a.k.a. RG1678, bitopertin) Hoffmann-La Roche | Single-center study will assess the effect on biomarkers measures of cognitive dysfunction, the clinical efficacy and, safety of RO4917383 (10 mg daily, orally) in patients with schizophrenia on stable antipsychotic medication | NCT01116830 | |
Phase II | PF-02545920 Pfizer | Assessed the efficacy and safety of an investigational compound PF-02545920 for the treatment of schizophrenia. PF-02545920 is expected to be more effective than placebo in reducing symptoms associated with schizophrenia | NCT00570063 Terminated 2008 |
PF-03463275 Pfizer | Examined the efficacy of PF-03463275 compared with placebo in treating negative symptoms of schizophrenia when added to ongoing antipsychotic treatment in stable outpatients with schizophrenia | NCT00977522 Terminated 2010 | |
SCH 900435 (a.k.a. Org 25935) Organon/Schering-Plough | Tested whether SCH 900435 (16 mg twice daily) is more effective than placebo in the treatment of patients with schizophrenia, using 15 mg olanzapine once daily as active control | NCT00988728 Terminated 2012 | |
Org 25935 (a.k.a. SCH 900435) Organon/Schering-Plough | Evaluated whether Org 25935 is more effective than placebo in improving negative symptoms in subjects with schizophrenia who are concurrently treated with a stable dose of a second-generation antipsychotic | NCT00725075 Completed 2008 | |
RO4917838 (a.k.a. RG1678, bitopertin) Hoffmann-La Roche | Evaluated the efficacy of RO4917838 (10, 30, or 60 mg) in patients with schizophrenia who are stable on current antipsychotic treatment (olanzapine, quetiapine, risperidone, paliperidone or aripiprazole) with prominent negative or disorganized thought symptoms | NCT00616798 Completed 2010 | |
Evaluated the efficacy of RO4917838 (bitopertin) in patients with acute exacerbation of schizophrenia. Patients receive either RO4917838 10 mg or RO4917838 30 mg or olanzapine 15 mg or placebo orally daily for 4 weeks as inpatients | NCT01234779 Completed 2014 | ||
Sarcosine Medical University of Lodz | To evaluate whether dietary supplement of sarcosine is effective in the treatment of schizophrenia with a focus on quality of life and sexual functioning | NCT01503359 | |
Sarcosine Chang-Hua Hospital, Taiwan | Efficacy and safety study of sarcosine as an adjunctive therapy for schizophrenia | NCT01047592 | |
Unspecified compound China Medical University Hospital | The study will investigate the efficacy and safety of NMDA adjuvant therapy (GlyT1 inhibition) in refractory schizophrenia, and identify the predictors for treatment response to NMDA enhancers | NCT01251055 | |
Phase III | RO4917838 (a.k.a. RG1678, bitopertin) Hoffmann-La Roche | Multi-center study assessing the efficacy of RO4917838 in schizophrenia patients with persistent negative symptoms on stable antipsychotic treatment. The study lasts for 52 weeks, followed by an optional treatment extension for up to 3 years | NCT01192867 NCT01192906 NCT01192880discontinued/completed 2014–5 |
Multi-center study assessing the efficacy and safety of RO4917838 in patients with sub-optimally controlled symptoms of schizophrenia. Patients, on stable treatment with antipsychotics, will be randomized to receive daily oral doses of RO4917838 or matching placebo for 52 weeks, followed by an optional treatment extension for up to 3 years | NCT01235520 NCT01235559 NCT01235585discontinued/completed 2014–5 | ||
It is not certain, however, if similar interactions with the dopamine neurotransmission are shared by other GlyT1 inhibitors. If so, this may predict weak efficacy against positive symptoms and provides a rationale for GlyT1 inhibition as an add-on to be combined with conventional antipsychotic drugs that are known to block dopamine D2 receptors [76–78]. On the other hand, the positive effects of SSR504734 on mesocortico-limbic dopamine may contribute to its efficacy to enhance working memory and problem solving in normal animals [79, 80] because stimulation of prefrontal dopamine activity could enhance cognitive performance in healthy humans and ameliorate cognitive deficits in schizophrenia patients [81–85]. However, this is unlikely the sole mechanism for the pro-cognitive potential of GlyT1 inhibitors. Several other GlyT1 inhibitors have also been shown to ameliorate the learning and memory deficits induced by NMDA receptor blockade across multiple tests (see Table 25.1).
The prepulse inhibition (PPI) and latent inhibition (LI) tests are two highly translational paradigms with face, construct, and predictive validity [86]. PPI measures a form of sensory gating that regulates stimulus access to higher cognitive resources. It is considered as a pre-attentional filtering mechanism whereby ongoing information processing is protected from intrusion by spurious environmental stimuli [87]. LI, on the other hand, refers to a specific form of selective attention whereby one learns to pay less attention to stimuli that are evidently irrelevant (i.e., lacking biological significance) in the past. This is an important component of associative learning, which suppresses the formation of spurious contingency between events in one’s environment, and thereby favors the learning of reliable contingency between environmental events to guide future behavior [88]. Schizophrenic patients exhibit deficits in PPI as well as in LI [89, 90]; and antipsychotic drugs can strengthen both phenomena [91–100]. With few exceptions, GlyT1 inhibitors exhibited antipsychotic potential in the PPI test (Table 25.1). All three compounds (ALX 5407, SSR504734, and SSR103800) that have been evaluated in the LI paradigm are effective in enhancing LI in normal animals [101, 102], as well as antagonizing the LI impairment induced by the NMDA receptor antagonist MK801—a specific form of LI deficit suggested to mimic attentional dysfunction linked to negative symptoms [103].
Additional tests that might point to an efficacy against affective symptoms in schizophrenia include enhanced social recognition memory by ALX 5407 [104], and antidepressant-like property in the Porsolt forced swim test by SSR103800 [105]. Indications for potential anti-anxiety effects of GlyT1 inhibitors include the suppression of contextual fear learning by SSR504734 [106] and the facilitation of fear extinction by ALX 5407 [101].
Proof of Principle I: Glycine-B Site Agonists
One of the first clinical trials evaluating the antipsychotic potential of glycine augmentation therapy was an open-label pilot study of orally administered glycine (10.8 g/day) as an add-on medication to conventional antipsychotics drugs [107]. The study yielded inconclusive results, and another study done by the same group using milacimide, a prodrug for glycine, failed to reveal any benefits [108].
These early negative findings were followed by a series of placebo-controlled clinical trials of glycine add-on therapy with substantially higher doses (0.4–0.8 g/kg/day) designed to overcome poor brain penetration of orally administered glycine. At the highest dose, which was associated with a six-fold increase in the plasma levels of glycine, glycine add-on therapy significantly ameliorated the negative symptoms and improved global functions in the patients. However, the required high doses are considered impractical for long-term clinical use [109] because of potential gastrointestinal disturbances [110, 111].
Subsequent add-on studies with the glycine-B site agonist, d-serine, revealed similar beneficial effects against the negative and cognitive symptoms at significantly lower doses (2–8 g/day) [112, 113]. Nephrotoxicity was a concern at the highest dose (8 g/day), but could be avoided at lower doses (≤4 g/day) that were still clinical effective [112]. As an alternative to overcome the poor brain penetrance of glycine, and to a lesser degree d-serine, the partial glycine-B site agonist, d-cycloserine, has also been evaluated as an add-on medication in clinical trials [114–118]. When added to conventional antipsychotics, d-cycloserine significantly reduced negative symptoms over a relatively small dose range with optimal therapeutic efficacy at 50 mg/day [114, 117, 119]. However, the clinical potential of d-cycloserine has been limited by the decrease in therapeutic efficacy over time, the modest effect size, and narrow effective dose range [116].
These studies have provided important evidence that adjunctive glycine augmentation therapy directed at increasing glycine-B site occupancy could improve negative and cognitive symptoms of schizophrenia. At the same time they have identified obvious limitations that justify the alternative approach to inhibit GlyT1. Targeting GlyT1 is an attractive strategy considering that the potential of designing new compounds that can selectively mimic the action of small molecules such as glycine and d-serine on the glycine-B site is limited because their simple backbone structures leave little room for possible structural modifications [113].
Proof of Principle II: GlyT1 Inhibition
The first proof-of-concept studies were carried out with the naturally occurring GlyT1 inhibitor, sarcosine (N-methyl glycine). Like glycine, sarcosine is a natural amino acid that is generated as an intermediate in the synthesis and degradation of glycine. Sarcosine is well tolerated and has no known toxicity as indicated by the lack of adverse effects in sarcosinemia, a rare congenital condition caused by dysfunction of sarcosine metabolism that leads to the accumulation of sarcosine in plasma and urine [120]. A number of randomized, double blind, placebo-controlled trials have shown that sarcosine add-on treatment improves positive and negative symptoms as well as general functions in patients stabilized on conventional antipsychotic medication [121–124]. The beneficial effects are not limited to patients with stable positive symptoms but are also seen in patients in the acute phase of the disease [122]. It should be emphasized that all these studies administered sarcosine as an add on, and so far only one small-scale non-placebo controlled trial had evaluated sarcosine as a monotherapy [124] and the tentative trend of negative symptoms reduction reported needs to be substantiated by standard controlled trials with larger sample size.
However, adjunctive sarcosine appeared ineffective when combined with clozapine [125], which is similar to what had been learned from combining glycine or d-serine with clozapine [126]. Adjunctive glycine treatment might worsen the positive symptoms in patients maintained on baseline clozapine [126], while a significant exacerbation of the negative symptoms has been observed when d-cycloserine is combined with clozapine [118]. The reason for the unique drug–drug interaction is not fully understood. It is suspected that clozapine may already increase synaptic glycine levels through as yet unknown mechanism [127]. Hence, baseline clozapine medication should be avoided; this has been recognized in all subsequent trials with synthetic GlyT1 inhibitors.
The encouraging outcomes with sarcosine have spearheaded the development of a variety of synthetic GlyT1 inhibitors. Many of them have been evaluated in clinical trials as a potential new class of antipsychotic drugs (Table 25.2). The first generation of synthetic compounds were substituted sarcosine derivatives characterized by irreversible and non-competitive inhibition of GlyT1 such as ALX 5407 and Org24589 [128]. They were associated with motor and respiratory side effects resulting from excessive glycinergic inhibition in the brain stem and cerebellum [128–130]. This has led to the development of a second generation of non-sarcosine-based compounds exhibiting reversible and competitive inhibition of glycine transport such as SSR504734 [128] which have been associated with fewer side effects. However, this view has been challenged by Kopec and colleagues [131] who contended that the induction of motor side effects may instead be better predicted by the target residence time, which refers to the dissociative half-life of the compound–target complex.
Related posts:
 Towards a Psychotherapy with a Philosophical Basis
Neurosciences, Neuroeconomics, and Metaphysics
Role of the Neuropeptide Angiotensin II in Stress and Related Disorders
Animal Models of Psychopathology and Its Relation to Clinical Practice
Adenosine in the Neurobiology of Schizophrenia: Potential Adenosine Receptor-Based Pharmacotherapy
Towards a Psychotherapy with a Philosophical Basis
Neurosciences, Neuroeconomics, and Metaphysics
Role of the Neuropeptide Angiotensin II in Stress and Related Disorders
Animal Models of Psychopathology and Its Relation to Clinical Practice
Adenosine in the Neurobiology of Schizophrenia: Potential Adenosine Receptor-Based Pharmacotherapy
 Attentional Capacity in Children: Intervention Programs for Its Development
Attentional Capacity in Children: Intervention Programs for Its Development
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


