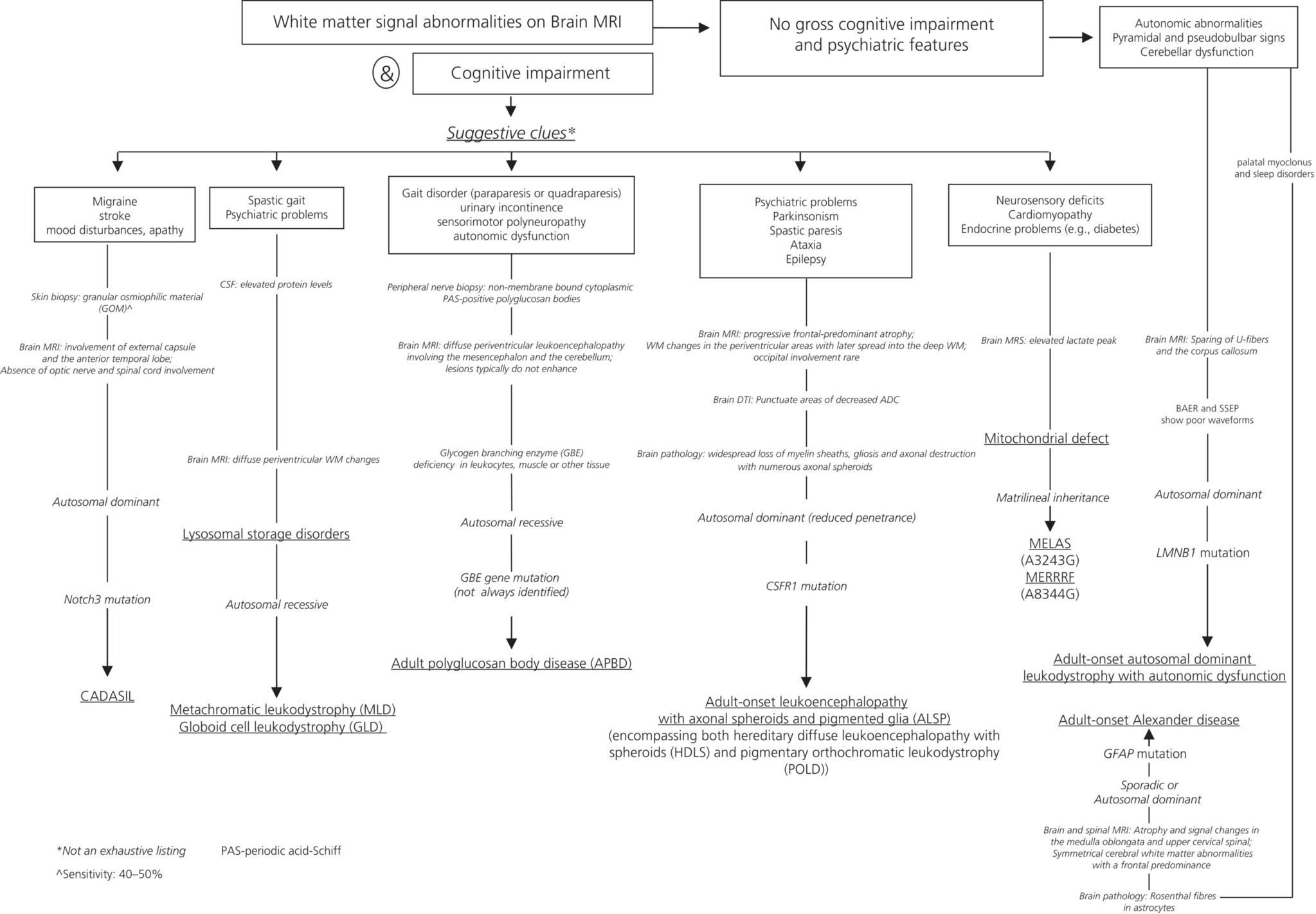
CHAPTER 12 Gregory M. Pastores1,2 and Swati A. Sathe3 1 Mater Misericordiae University Hospital, Dublin, Ireland 2 Yale University School of Medicine, New Haven, CT, USA 3 Rutgers, The State University of New Jersey, New Jersey Medical School, Newark, NJ, USA The leukoencephalopathies encompass a group of heterogeneous disorders that can lead to cognitive problems as a consequence of brain white matter involvement. Many of the acquired leukoencephalopathies that cause cognitive decline such as inflammatory, vascular, infectious, neoplastic, and toxic diseases are covered elsewhere in the book. Our focus will be on the hereditary forms of white matter disorders, that is, leukodystrophies, in particular the adult-onset conditions leading to dementia as a predominant feature (Table 12.1). Strictly, the term leukodystrophy is applied to conditions which have a genetic basis, progressive course, and white matter involvement (Table 12.2) [1]. In several, the biochemical and/or molecular defect is known, enabling diagnostic confirmation, which then allows appropriate genetic counseling and prognostication. Table 12.1 Selected leukodystrophies. Table 12.2 Clinical signs of a leukodystrophy. The leukodystrophies are familiar to most in their so-called classic expression with onset usually in childhood. It must be recognized that there are late-onset presentations of almost every childhood leukodystrophy. In such cases, disease course is often insidious, and diagnosis can be significantly delayed. It is not uncommon for patients with late-onset leukodystrophy to be initially suspected and treated as primary or secondary progressive multiple sclerosis (MS) [2]. Given the complex clinical presentations and similarity with more common adult-onset white matter diseases, underdiagnosis of leukodystrophy is likely. A negative family history or low disease prevalence often complicates this issue. Review of systems and laboratory assessments in patients with a leukodystrophy often show no indication of electrolyte imbalance, thyroid or liver disease, vitamin deficiency, brain mass, drug intoxication, or chronic infection. In most cases, cerebrospinal fluid (CSF) studies, including cell counts, glucose, and oligoclonal bands, also are normal. In metachromatic leukodystrophy (MLD) or globoid cell leukodystrophy (GLD), however, there is increased CSF protein levels; the diagnosis of these two lysosomal disorders can be confirmed by biochemical enzyme testing (discussed in the following) [3]. Although certain MRI features in leukodystrophy are quite characteristic, they might also suggest several alternate diagnoses. Those include white matter diseases such as MS, cerebrovascular diseases (e.g., Binswanger’s disease), or chronic exposure to toxic substances (e.g., chemotherapy). The pattern and distribution of white matter changes on MRI in patients with a leukodystrophy might suggest a particular disorder or narrow the list of differential diagnoses [4]. Until proven otherwise, symmetrical changes consistent with white matter disease is indicative of a leukodystrophy. The differential diagnosis of a leukodystrophy can also be aided by incorporating the results of a neurological evaluation and genetic testing (see Table 12.3 for a review of clinical and MRI features associated with the disorders reviewed in this chapter). Figure 12.1 provides a framework for the clinical workup of a patient with suspected leukodystrophy or leukoencephalopathy. Table 12.3 Key features of leukodystrophies and leukoencephalopathies discussed in this chapter. AD, autosomal dominant; ALS, amyotrophic lateral sclerosis; ALSP, adult-onset leukoencephalopathy with axonal spheroids and pigmented glia; AMN, adrenomyeloneuropathy (form of X-ALD); AR, autosomal recessive; ARSA, arylsulfatase A; CC, corpus callosum; CSO, centrum semiovale; EC, external capsule; EP, extrapyramidal symptoms; ERG, electroretinogram; GALC, galactocerebrosidase; GBE, glycogen branching enzyme; GFAP, glial fibrillary acidic protein; IC, internal capsule; NCS, nerve conduction studies; PBs, polyglucosan bodies; PVWM, periventricular white matter; RLS, restless legs syndrome; SIVD, small vessel ischemic vascular disease; SSEPs, somatosensory evoked potentials; TIAs, transient ischemic attacks; VEPs, visual evoked potentials; VLCFAs, very-long-chain fatty acids; WM, white matter. Figure 12.1 Flow chart listing selected leukodystrophies, their characteristic features, and mode of inheritance. The management of patients with most leukodystrophies currently unfortunately is primarily symptomatic. Hematopoietic stem cell transplantation and pharmacologic treatments are options in selected cases. For disorders that are potentially treatable, early diagnosis is critical for the best possible outcome. A 41-year-old gentleman was seen because of migraine with aura since age 24 and multiple recurrent episodes of unilateral numbness and tingling, attributed to MS. Regarding the migraines, his visual auras were reported as bright spots and lines. Some episodes of headache were associated with confusion which typically occurred about half an hour after the onset of headache. During the headache, he is unable to recognize people he knows and does not fully comprehend what is being said to him, his speech is affected, and he has word-finding difficulty. His problems usually resolve within hours, and he has partial memory for the episode. At age 35, he suffered from an acute episode of numbness and clumsiness in the left face and arm with a field cut, with complete recovery. A brain MRI done at the time showed extensive white matter disease. He was diagnosed with MS after excluding HIV, Lyme disease, and lupus. He was treated with glatiramer acetate for 1 year without benefit, following which he was given interferon beta-1 for another year, again without clear benefit. Over the past 6 years since the first episode, he has had four additional episodes of left-sided numbness and clumsiness with left field cut. Over the past 10–15 years, there has been a gradual decline in his memory and concentration. He keeps a diary to remember appointments and his schedule. Additionally, he has to pay close attention to his daily work routine as a machine operator to ensure that he correctly follows the sequence. There are also subtle changes in his personality such that he is more emotional and cries easily. His past medical history revealed major depression with suicidal ideation at age 24, elevated triglycerides (treated successfully with niacin), and no history of hypertension/diabetes/asthma or heart disease. His family history was remarkable for one older brother with migraine and episodes of arm and face numbness, attributed to MS. His father died at age 42 from bone cancer, and his mother, age 65, has hypertriglyceridemia, osteoarthritis, and cervical spondylosis. She has no history of migraine, stroke, or dementia. On neurological examination, he was alert and oriented with fluent speech. Comprehension, repetition, and naming were intact. Three object recall was 2/3 (3/3 with cues). Neuropsychological testing showed impairment in attention, processing speed, and executive function, including deficits in timed measures and measures of error monitoring, but with spared memory. Cranial nerve, motor, and sensory examinations were normal. There was no pronator drift. Rapid alternating movements were accurate. There was no double simultaneous extinction. Deep tendon reflexes were bilaterally symmetric, 2+. Both plantars were flexor. Gait was normal and there were no cerebellar signs. His clinical course, brain MRI findings (Figure 12.2), and family history were deemed consistent with cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). The absence of optic nerve and spinal cord involvement on brain MRI, the absence of oligoclonal bands in the CSF, and the absence of hypertension are pertinent negative findings that help to distinguish cases of CADASIL from MS, small vessel disease, or primary angiitis of the nervous system. A significant proportion of patients with CADASIL often are initially diagnosed and treated for MS. Thus, evaluation of patients suspected to have MS should include careful consideration of red flags, which are features in the history, examination, or diagnostic tests that are not typical or suggestive of MS and may point toward the diagnosis of a leukodystrophy [5]. Figure 12.2 FLAIR brain MRI in a 50-year-old with CADASIL. Some classic MRI findings of CADASIL including T2-weighted medial temporal lobe hyperintensities (solid arrow), cavitations in the white matter (dashed arrows), and confluent white matter disease are shown. CADASIL is included in this chapter although it is not a leukodystrophy in the conventional sense, but a vascular disease. Van Bogaert’s description in 1955 of “Binswanger’s disease with a rapid course in two sisters” probably depicts the first description of CADASIL. The acronym stands for cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, which essentially describes the key features of the disorder (i.e., hereditary small vessel disease leading to strokes and dementia). The overall prevalence of CADASIL is unknown, but one small study from Scotland provided an estimate of 4.14 cases per 100 000 [6]. Several main symptoms are described in CADASIL including migraine with aura, ischemic events (stroke), seizures, episodes of confusion (unexplained neurological episodes), mood disturbances, apathy, and cognitive impairment [7]. These symptoms occur in a successive manner over decades. Extraneurologic symptoms usually do not occur with CADASIL; this contrasts with Fabry disease (another entity associated with brain white matter changes, often erroneously attributed to MS). In Fabry disease, patients often develop renal insufficiency and cardiomyopathy prior to onset of the central nervous system (CNS) disease (discussed in the following). Forty to 50% of patients with CADASIL have migraine with aura, which is usually the first symptom, with onset on average at age 30 years (range 6–48 years) [7]. The most common auras are visual and sensory; however, 50% of patients also have atypical attacks with basilar, hemiplegic, or prolonged aura, and some patients have very severe attacks with confusion, fever, meningitis, or coma. Sixty to eighty five percent of patients with CADASIL suffer transient ischemic attacks (TIA) and ischemic strokes in the absence of other risk factors, at a mean age of 49 years (range 20–70 years) and at an estimated incidence of 10 episodes per 100 patient-years [7]. Ischemic events are almost invariably subcortical presenting as lacunar syndromes, for example, pure motor or sensory deficit, ataxic hemiparesis, and clumsy hand–dysarthria syndrome. Recurrent strokes over the years lead to significant morbidity with motor weakness, spasticity, gait difficulties, urinary urgency with or without incontinence, and pseudobulbar palsy. Depressive episodes are described in 20% of cases, which may alternate with manic episodes [8]. Independent of depression, apathy is seen in about 40% of cases [9]. Cognitive impairment, the second most frequent clinical manifestation after migraine, follows a distinctive pattern of neuropsychological abnormalities similar to subcortical ischemic vascular dementia [10]. Executive dysfunction and attentional deficits are frequently impaired early in the course of the disease, often occurring as early as 35–50 years of age [11]. Cognitive dysfunction is accompanied by a narrowing range of interests. Patients with an overt stroke fare worse on tests of executive dysfunction [12]. Prominent areas of deficit are processing speed, for example, in timed measures of the Trail Making Test parts A and B (TMT-A and TMT-B) and a symbol digit test [10–12]. In patients with both early and advanced disease, TMT-B is abnormal, indicative of problems with processing speed and set shifting. Deficits are recorded frequently in verbal fluency, which has a strong executive component, especially in letter fluency more so than category or semantic fluency. Similarly, impairment in error monitoring on neuropsychological testing emphasizes the executive deficit [10–12]. On memory testing, there is often mild or worse impairment on both immediate and delayed free recall, but cued recall and recognition are often intact. This suggests that the encoding component of memory is relatively preserved. Memory problems are usually due to retrieval deficits, as is commonly found in small vessel disease-induced vascular cognitive impairment [12]. Visuospatial functions and abstract reasoning are largely preserved, particularly during early disease stages. Lacunar infarcts, cerebral microbleeds, hippocampal volume, and brain atrophy are all shown to be independently associated with executive dysfunction when adjusted for age [13]. White matter hyperintensities appear to be less directly related to the cognitive decline. Cognitive decline progresses with age and at late stages involves verbal or visual memory, language, reasoning, and visuospatial abilities [10]. Severe aphasia, apraxia, or agnosia is rare. Seizures, intracerebral hemorrhages, territorial infarcts (possibly coincidental), and extrapyramidal features are rarely reported [7]. Typically, punctiform or nodular T2/FLAIR hyperintensities in periventricular areas and in the centrum semiovale are the first sign on MRI, which progress over decades to diffuse, extensive, and somewhat symmetrical hyperintensities, often with cavitations [7, 10]. White matter abnormalities probably precede the onset of clinical symptoms by at least 10–15 years; in any case, the brain MRI abnormalities are consistently seen by the age of 35. The frequency and severity of these T2 white matter abnormalities progress with age. MRI involvement of the external capsule and the anterior part of the temporal lobe is characteristic of CADASIL, helping to differentiate it from MS [7, 10]. Lacunar infarcts, which are often punctate or larger in size, appear later in life as T1 hypointense lesions in the same areas as the T2 signal changes [13]. Recent infarcts (within the past ~10 days) show hyperintensity (restricted diffusion) on diffusion-weighted MRI. With advancing age, microbleeds are commonly seen on gradient echo images. On diffusion tensor imaging (DTI), increased water diffusion is seen in the thalamus that may not be observed on standard T2/FLAIR neuroimaging. These DTI abnormalities are more strongly correlated with measures of executive dysfunction and clinical disability in comparison to T2 hyperintensities. Thus, diffusion histograms might be used as a marker of disease progression. In addition to white matter findings, global brain atrophy progresses three times more rapidly in patients with CADASIL compared to normal aging, and the extent of brain atrophy correlates with cognitive impairment and disability scales [14]. Macroscopic neuropathological examination of the brain shows diffuse myelin pallor and rarefaction of white matter in periventricular areas and centrum semiovale, lacunar infarcts in white matter and basal ganglia, and dilated Virchow–Robin spaces [7, 10]. Neuronal apoptosis in the cortex correlates with the extent of subcortical ischemic lesion load. Microscopically, the arteriopathy, characterized by thickening of the arterial wall causing luminal stenosis, deposition of nonamyloid granular osmiophilic material (GOM) in the media extending into the adventitia, and eventual disintegration of smooth muscle cells, is found in the small penetrating cerebral and leptomeningeal arteries. This in part explains the unusual pattern of white matter abnormalities seen in CADASIL. The endothelium is largely normal. Extracellular GOM, located close to the cell surface of smooth muscle cells, is pathognomonic of CADASIL. Arteriopathy is found in other organs, such as the spleen, liver, kidneys, muscle, aorta, and skin, although clinical manifestations are restricted to the CNS. Therefore, a skin biopsy sample demonstrating this pathologic change is used for diagnosis; the sensitivity of this test is 40–50% [7, 10]. Immunostaining with NOTCH3 monoclonal antibody to detect the accumulation of NOTCH3 protein in the vessel wall is highly sensitive (85–95%) and specific (95–100%) [7, 10]. CADASIL, caused by mutations in the Notch3 gene encoded on chromosome 19q12, is inherited in an autosomal dominant manner. The Notch3 gene encodes a single-pass transmembrane receptor of 2321 amino acids with an extracellular domain containing 34 epidermal growth factor repeats (EGFR). Each EGFR has six cysteine residues. More than 95% of the 150 mutations described thus far are missense mutations found in exons 2–24, and that lead to addition or deletion of a cysteine residue in the EGFR [7, 10]. De novo mutations and homozygous mutations are rarely reported. Molecular testing by screening exons 2–24 is the gold standard for the diagnosis of CADASIL; the test is 100% specific when a mutation involving cysteine residue is detected, and the sensitivity is close to 100%. In asymptomatic adult first-degree family members of patients with CADASIL, genetic testing raises psychological and ethical concerns similar to other adult-onset autosomal dominant neurodegenerative disorders such as Huntington’s disease. There is no recommendation to screen children, as currently there is no benefit in terms of treatment. NOTCH3 is predominantly expressed in vascular smooth muscle cells of small arteries in particular. CADASIL mutations cause gradual accumulation of the extracellular domain of NOTCH3 protein in the form of microscopic aggregates around vascular smooth muscle cells and pericytes of brain arteries and capillaries, in close proximity to deposits of GOM [15]. Total loss of Notch3, however, does not cause CADASIL pathology. Evidence strongly suggests that CADASIL mutations act through gain of novel function mechanisms and that the change in the number of cysteine residues in NOTCH3 is the common denominator which affects the survival and function of vascular smooth muscle cells [15]. Chronic subcortical ischemia and compromised cerebral hemodynamics in addition to altered vasoreactivity resulting from structural and functional changes in brain arteries might lead to recurrent lacunar infarcts and microstructural alterations that ultimately cause cognitive decline, motor disability, cortical atrophy, and neuronal apoptosis [13]. The pathophysiology of mood disorders and migraine with aura is mostly unknown. Cortical morphology (i.e., depth and width of cortical sulci) in the mediofrontal and orbitofrontal areas, in contrast to cortical thickness, has been strongly and independently associated with apathy in CADASIL [16]. One recent study suggests that increased rapid-onset cortical plasticity may contribute to largely preserved cognitive and motor function in patients with CADASIL despite extensive ischemic small vessel disease [16]. At present, there is no treatment for CADASIL. Migraine with aura is treated similarly to migraine in general population, with the exception that ergot derivatives and triptans are not considered safe in views of their vasoconstricting property [7, 10]. Usual prophylactic drugs such as antiepileptic drugs, tricyclic antidepressants, or antihypertensives can be used. Anecdotally, acetazolamide has been reported to be effective. Secondary stroke prevention similar to noncardioembolic ischemic stroke is usually recommended: use of antiplatelet drugs and treatment of vascular risk factors. Use of anticoagulants is avoided because of the increased risk of intracerebral hemorrhage in the presence of cerebral microbleeds. Donepezil was tested in CADASIL patients with cognitive impairment. Inclusion criteria were a Mini-Mental State Examination score of 10–27 or a TMT-B time score that is at least 1.5 SD below the mean, after adjustment for age and education [17]. One hundred sixty-eight patients were followed for 18 weeks. No effect on the primary endpoint, using the cognitive subscale of the vascular Alzheimer’s disease cognitive assessment scale (ADAS-Cog), was noted; improvements, however, were recorded on measures of executive functions [17]. Supportive measures such as physical therapy and rehabilitation, psychological support, and nursing care play an important role in the long-term management of elderly debilitated and demented individuals. Genetic counseling for asymptomatic members at risk of carrying the mutation is vital prior to testing.
Leukoencephalopathies/leukodystrophies
Introduction
Disorder
Cause
Disease
Age at (adult) onset
Key clinical features
Cognitive profile
Psychiatric findings
Distinguishing MRI features
Genetics
Diagnosis
CADASIL
30–50s; milder cases in 60s
Stroke/TIAs, migraines, confusional episodes, seizures
Slowed processing speed, frontal-executive deficits
Apathy, depression, lability
AD
Notch3 gene mutation, GOM on skin biopsy with or without Notch3 immunostaining
HDLS (ALSP)
39 (±15 SD)
Dementia, stiffness, clumsiness, weakness, ataxia, extrapyramidal, and/or pyramidal dysfunction
Frontal syndrome with memory impairment, intellectual deterioration.
Often initial symptoms, depression, apathy, and blunting
AD (reduced penetrance)
Clinical features, MRI/MRS, and brain pathologyCSF1R gene mutation
Rapid-onset cases reported
Normal NCS, VEPs, SSEPs, and ERGs
POLD (ALSP)
43 (±13 SD)
Dementia, seizures, pyramidal (spastic paresis), ataxia, dysphagia, dysarthria. Normal NCS, VEPs, SSEPs, and ERGs
Frontal-executive
Mood disorder, apathy, disinhibition, aggressiveness, and euphoria; might present like Pick’s. Psychosis less common
AD (reduced penetrance; possibly some sporadic cases)
Clinical features, MRI/MRS, and brain pathology
APBD
40s–60s
Tetrad: Urinary freq/urgency, gait disorder (spasticity), sensory > motor polyneuropathy, cognitive impairment. Less common EP, cerebellar ataxia, ALS, and cardiomyopathy. NCS – sensorimotor polyneuropathy
Dementia in approximately 60%; when present, involves memory loss
Not described. Possible depression
AR, rare sporadic.
Sural n. biopsy PBs in myelinated axons, decreased GBE in WBCs and cultured fibroblasts, mutation(s) in GBE gene
ADLD
30s–40s
Early autonomic dysfunction, pyramidal (spasticity) and pseudobulbar signs, and cerebellar dysfunction such as action tremor
Absence of gross impairment
Absence of profound psychiatric features
AD (highly penetrant)
Duplication of LMNB1 gene.
NO peripheral neuropathy. Normal NCS, VEPs. SSEPs abnormal. Autonomic skin innervation
Adult-onset Alexander Disease
Teens to 60s
Common: pyramidal tract signs (spasticity and hyperreflexia), cerebellar signs (ataxia, nystagmus, dysmetria), urinary symptoms, bulbar/pseudobulbar signs (palatal myoclonus and dysphagia, dysphonia)
Frontal-executive, memory
Not described
Usually sporadic; rare AD
GFAP gene mutation
Less common: dysautonomia, sleep apnea and RLS; poor fine motor skills.
SEPs, VEPs, and BAERs often abnormal
MLD (adult onset)
(Adult form) Teens to 40s–50s.
Elevated CSF protein
Progressive mental deterioration
Behavior/mood
Symmetric, confluent, T2 hyperintense WM, demyelinating appearance, often sparing of U-fibers
AR
Elevated urine sulfatide or ARSA gene mutation
Peripheral neuropathy (might be subclinical in late-onset forms)
GLD/Krabbe (adult onset)
Variable; Includes those with subtly earlier symptoms, but diagnosed as adults versus adult onset after age 20, as late as 60s
Elevated CSF protein
Progressive mental deterioration
Behavior/mood
Symmetric, confluent, T2 hyperintense WM, severe demyelination
AR
Very low GALC enzyme activity in leukocytes isolated from whole heparinized blood and cultured skin fibroblasts. In carriers, GALC assay often WNL; test for GALC gene mutation. Most common mutation in adult-onset Krabbe is c.857G>A mutation
Peripheral neuropathy (might be subclinical in late-onset forms)
Fabry
Teens to approximately 40s
Renal insufficiency, cardiomyopathy. Corneal and lenticular opacities
Might occur poststroke
Depression
Consistent with ischemic vascular disease: PVWM disease, WM signal intensity abnormalities and single or multiple lacunar infarcts, large ischemic cerebral infarctions. Posterior thalamus involvement (pulvinar sign—T1 bright) might occur
X linked
Deficiency of the lysosomal hydrolases α-galactosidase A (AGAL); in females, AGAL might be normal, so need GAL gene mutation
Strokes, SIVD
Acroparesthesias/pain might occur
Tortuosity and dilatation of the larger vessels
Female carriers often affected; variable presentation
X-ALD (adult onset)
Variable; AMN in 20s–middle age; female carriers might present >35 years old with mild disease
Spastic gait
Subacute decline. Cognitive deficits correlate with MRI findings. Might present as FTD
More common in childhood forms
Either caudorostral WM progression starting from initial parieto-occipital
X linked
Elevated VLCFAs (tissues, serum, and other fluids)—might be WNL in females, so check for ALDP gene mutation
Involvement beginning in the splenium of CC (65% of cases) or a rostrocaudal progression starting frontally beginning in the genu of CC (35% of cases). Tract involvement in the corticospinal, spinothalamic, visual, and auditory pathways
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
Illustrative case history 1
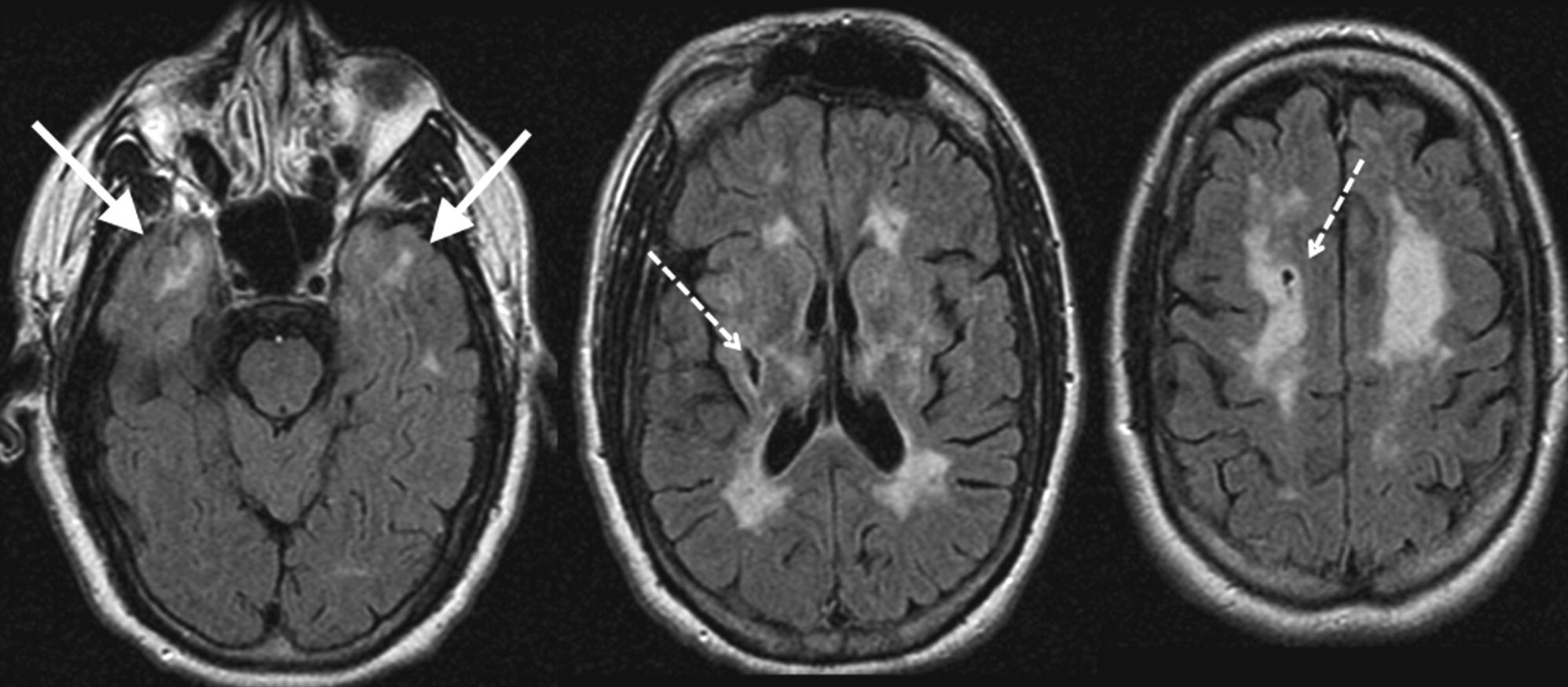
Background
Clinical presentation
Imaging
Pathology
Molecular genetics
Mechanisms underlying symptoms
Treatment
Adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: Hereditary diffuse leukoencephalopathy with spheroids and pigmentary orthochromatic leukodystrophy
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


