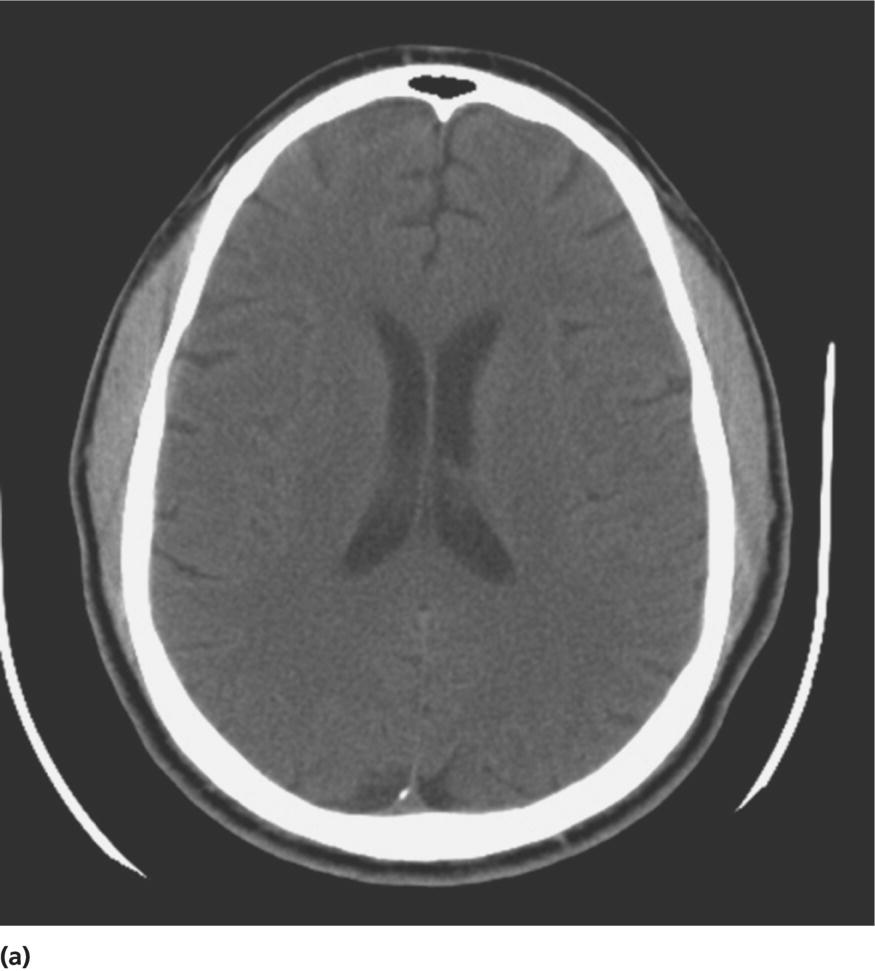
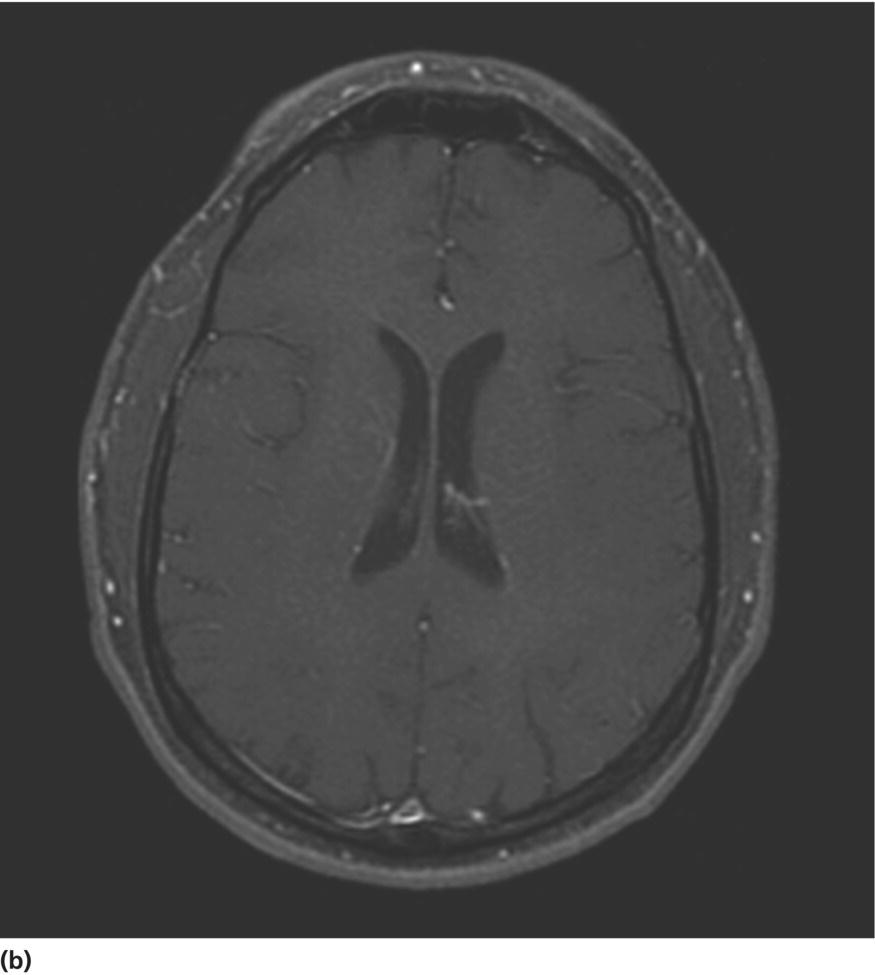
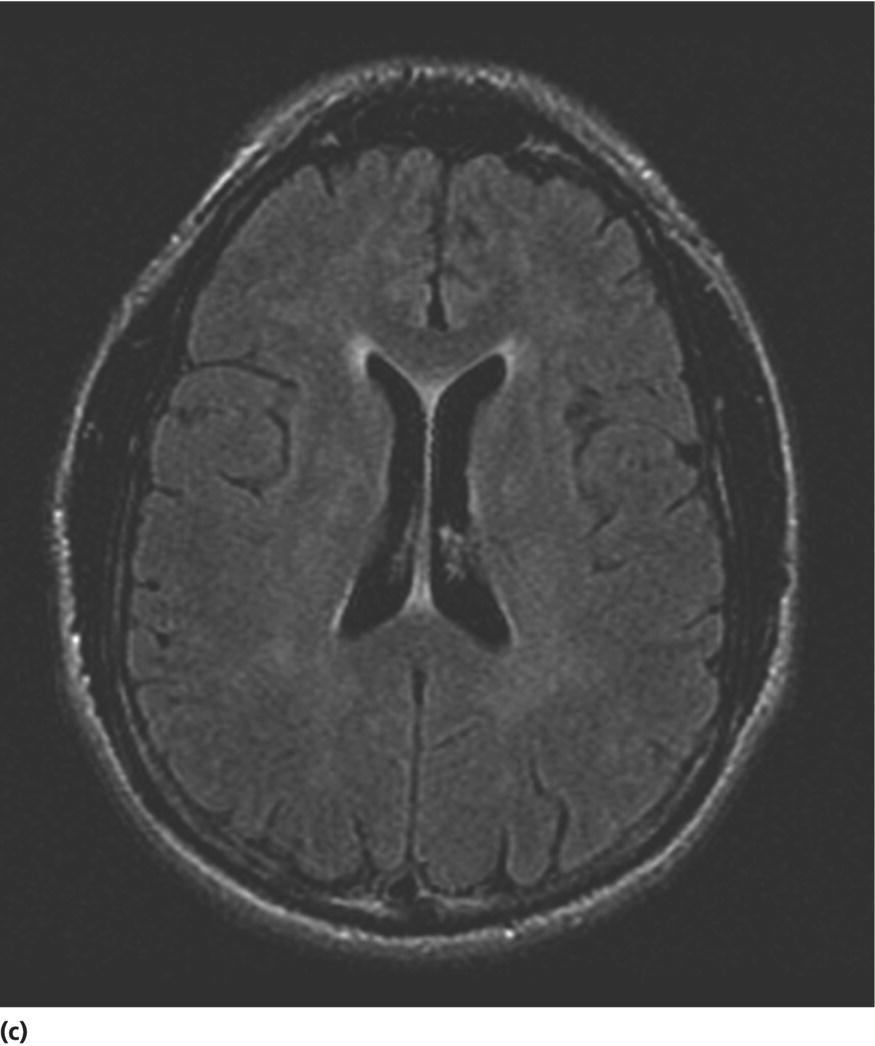
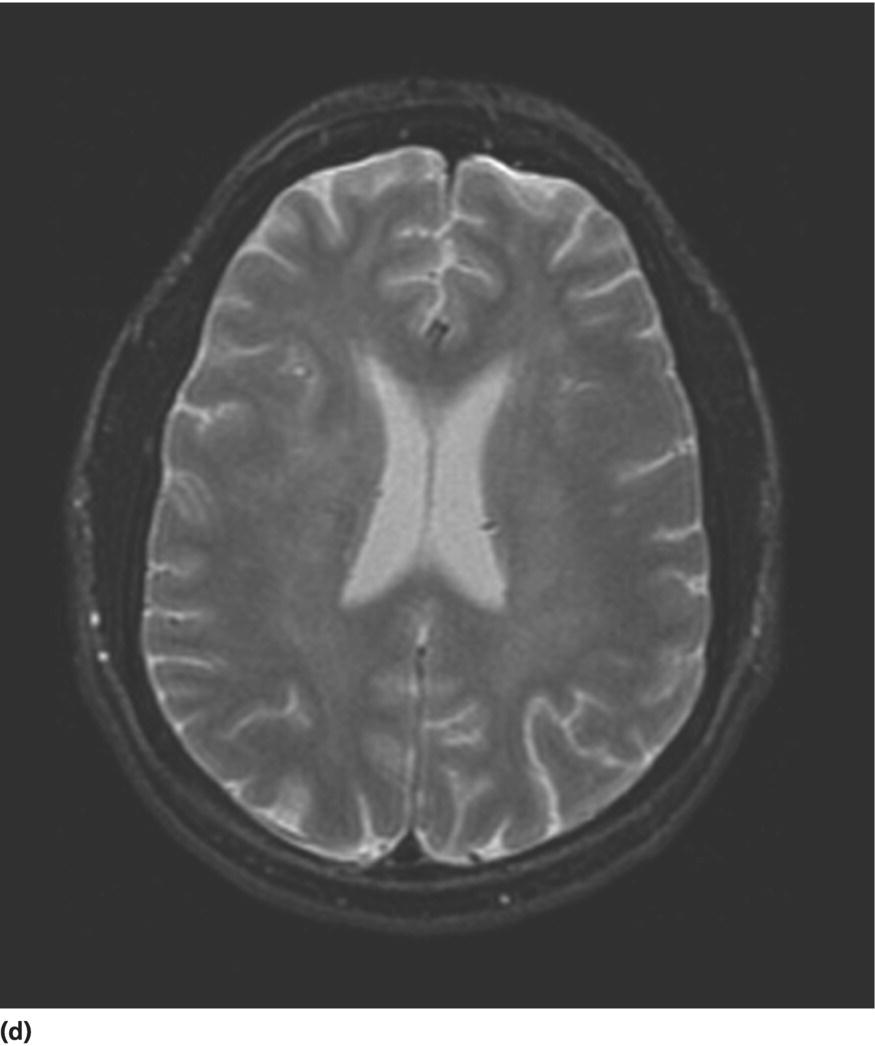
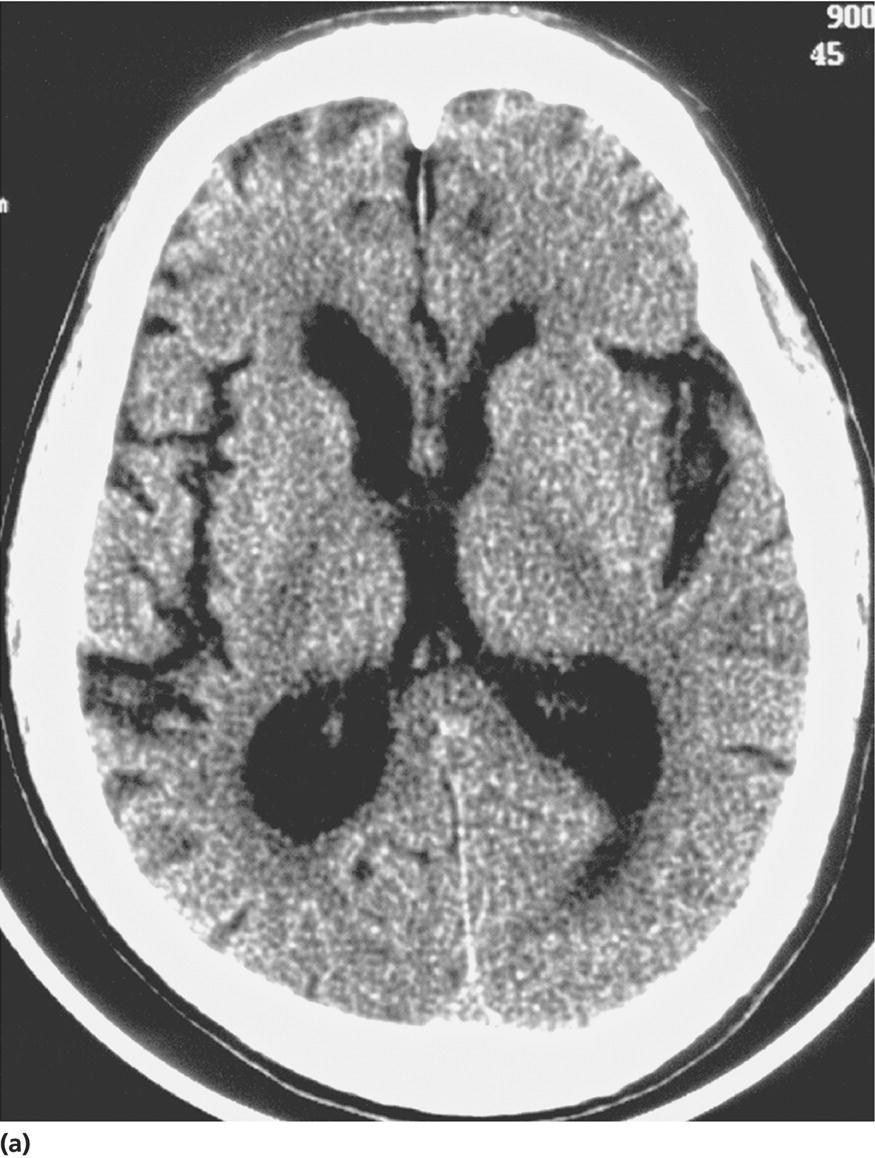
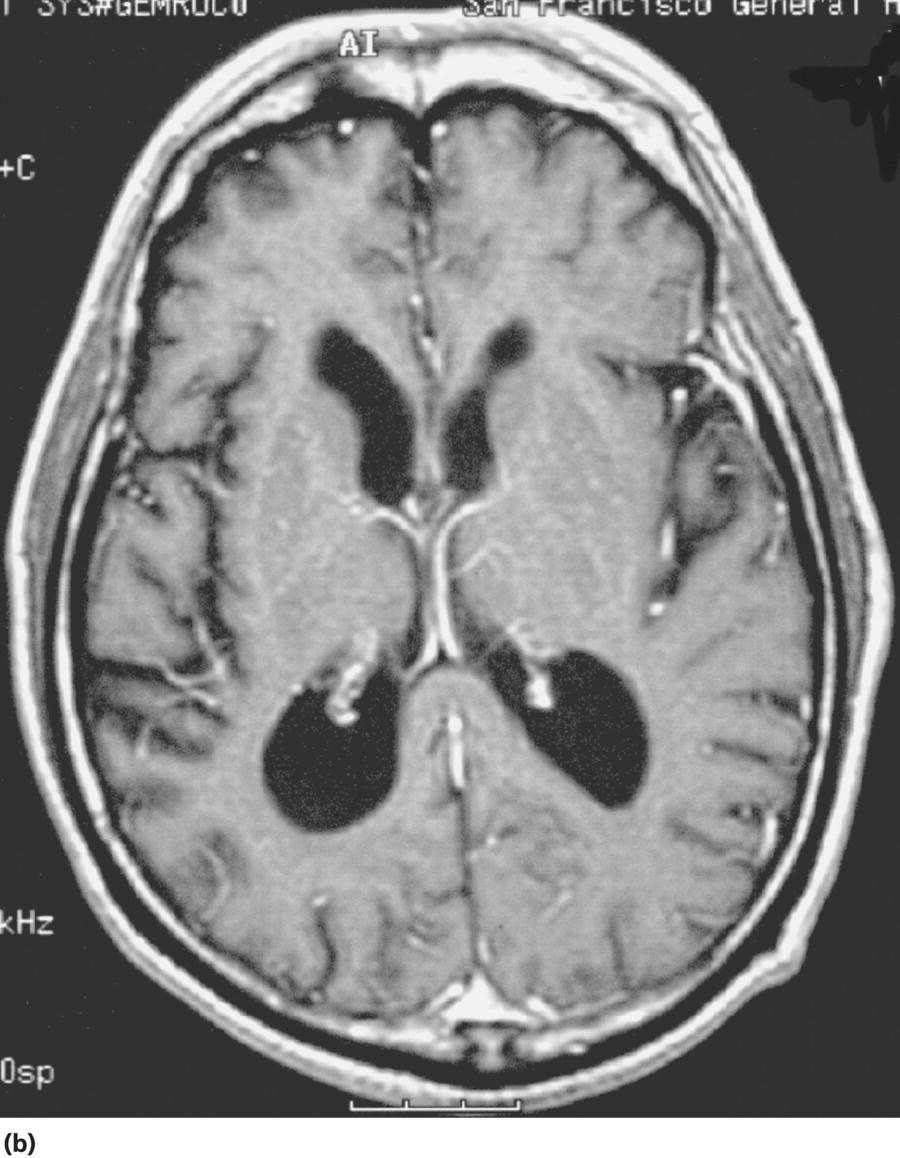
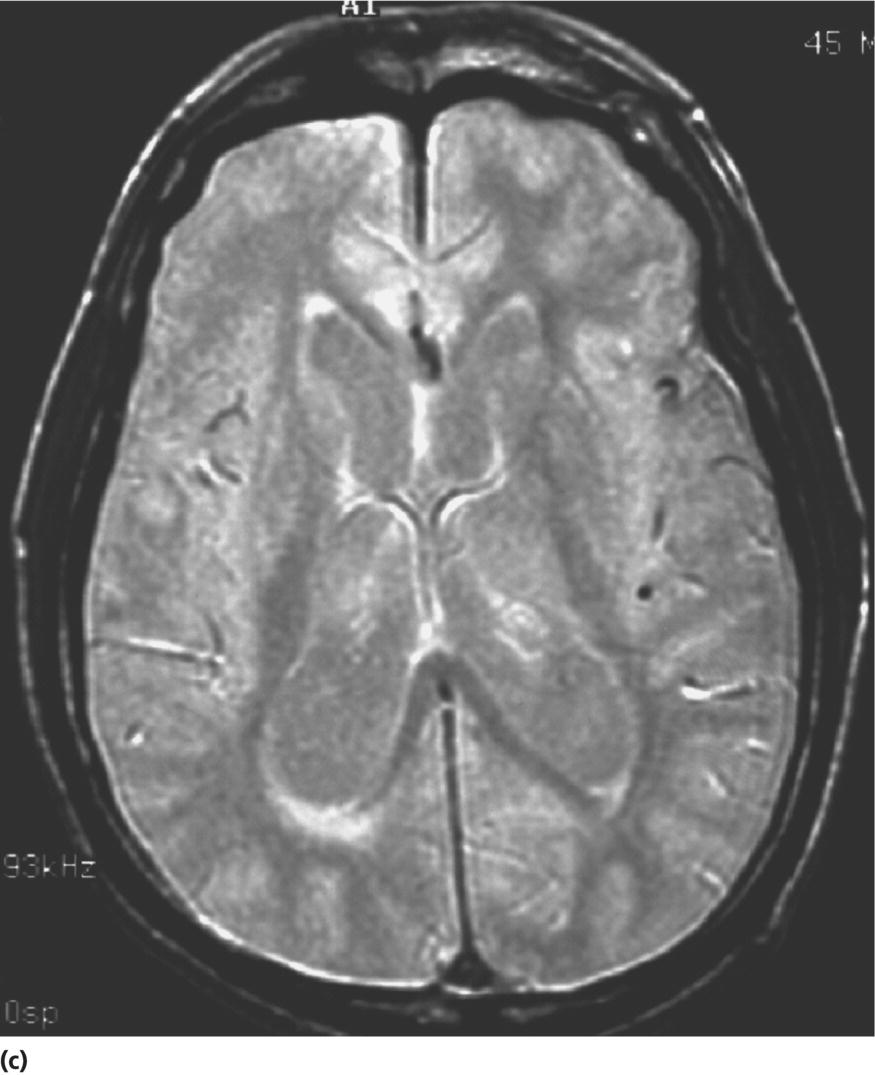
CHAPTER 13 Cheryl A. Jay1,2, Emily L. Ho3,4 and John Halperin5,6,7 1 University of California, San Francisco, San Francisco, CA, USA 2 San Francisco General Hospital (SFGH), San Francisco, CA, USA 3 University of Washington, Seattle, WA, USA 4 Swedish Neuroscience Institute, Seattle, WA, USA 5 Atlantic Neuroscience Institute, Summit, NJ, USA 6 Sidney Kimmel Medical College of Thomas Jefferson University, Philadelphia, PA, USA 7 Overlook Medical Center, Summit, NJ, USA Viruses, bacteria, fungi, and parasites most commonly reach the brain hematogenously during disseminated infection, although a few pathogens target specific neuronal or glial cells. Acute cerebral infections, such as community-acquired bacterial meningitis and most viral encephalitides, might begin with cognitive and personality dysfunction, but the rapid deterioration of alertness over subsequent hours to days causes little diagnostic confusion with most neurodegenerative disorders. Neuropsychological dysfunction is often a long-term, disabling complication of many acute brain infections. Patients with subacute or chronic infections develop cognitive and behavioral disturbances in several settings. A few nonprion chronic cerebral infections, such as HIV-associated dementia (HAD) and general paresis, manifest primarily as either a rapidly progressive or a more indolent dementia. Memory and behavioral complaints, with or without abnormalities by neuropsychological testing, are common among patients with some chronic cerebral or systemic infections in which other clinical features predominate. Neuropsychological dysfunction often is the predominant feature in the early weeks and months of other nonacute brain infections, including the long list of pathogens that cause chronic meningitis. The general approach to such patients is beyond the scope of this chapter and has been the subject of reviews [1, 2]. The focus here will be infections whose manifestations overlap with neurodegenerative disorders, along with briefer discussions of the neuropsychological sequelae of acute cerebral infections. Case: A young man with AIDS for 8 years and previously treated neurosyphilis presented to a clinic for evaluation of memory and gait difficulties for several weeks, on a background of a decreasing CD4+ T-cell count in the past year. He was not on antiretroviral therapy because of poor adherence related to active methamphetamine use. His physician noted him to be less jovial and talkative than usual. On examination, he had impaired reverse digit span and short-term memory. Reflexes were symmetrically diminished in the legs, with flexor plantar responses bilaterally. There was length-dependent sensory loss to the knees and wrists. Although Romberg’s test was negative, he walked on a slightly broad base with impaired tandem gait. Laboratory studies revealed a low CD4+ T-cell count at 98 cells/mm3 (normal = 500–1500 cells/mm3) with HIV viral load of greater than 200 000 copies/mL. Serum rapid plasma reagin (RPR) serology was 1:2 (1:1024 2 years previously prior to the treatment for neurosyphilis). CT was normal. MRI showed symmetric periventricular white matter abnormalities, compatible with HIV infection (Figure 13.1). CSF showed mildly elevated protein of 77 mg/dL (normal 15–45 mg/dl), glucose of 56 mg/dL, 7 RBC/mm3, and a slight pleocytosis of 12 WBC/mm3 (91% lymphocytes), with negative cryptococcal antigen and Venereal Disease Research Laboratory (VDRL) test. He was presumptively diagnosed with HAD, began combination antiretroviral therapy (cART), and was referred to drug rehabilitation (follow-up continued below). Figure 13.1 Neuroimaging in HIV-associated dementia studies obtained from a patient with untreated HIV infection, cognitive impairment, and gait disorder (see text). Noncontrast head CT (a) and T1-weighted brain MRI after gadolinium administration (b) are normal. FLAIR (c) and T2-weighted (d) images demonstrate ill-defined, symmetric white matter abnormalities, consistent with HIV-associated dementia. Soon after outbreaks of opportunistic infections (OIs) in Los Angeles among men who have sex with men heralded the HIV/AIDS pandemic in 1981, the nervous system emerged as a major site of clinical involvement [3]. Manifestations included cerebral OIs and neoplasms from severe immunocompromise, peripheral neuropathy, and a dementing illness initially referred to as subacute encephalitis [3–5]. The clinical syndrome, termed AIDS dementia complex (ADC) [4], and its neuropathologic substrate [5] were defined further in 1986. HIV encephalopathy [6] and AIDS-related dementia [7] were other terms for the occasionally static but more characteristically progressive cognitive, motor, and behavioral impairments that characterized the syndrome before cART [4, 6, 7]. Typical early symptoms include apathy, slowed thinking, impaired memory, and gait difficulty; neurological examination might be normal or reveal psychomotor slowing, hyperreflexia (sometimes masked by concomitant distal symmetric polyneuropathy), and pathological reflexes [4, 7]. In untreated patients, these early features often progress over months to mutism, quadriparesis, and incontinence [4]. Occasional patients may present with psychosis or mania [4, 7]. The Memorial Sloan Kettering (MSK) scale for ADC (Table 13.1), a staging system devised for patient care and clinical research in the late 1980s [8], remains in use. Host risk factors for dementia in the pre-cART era included low CD4 count, hemoglobin, or body mass, as well as constitutional symptoms, advancing age, and elevated cerebrospinal fluid (CSF) HIV viral load [9, 10]. Before cART, dementia was common, affecting approximately half of the HIV-infected patients [11]. Table 13.1 Memorial Sloan Kettering scale for AIDS Dementia Complex (ADC). Source: Price and Brew [8]. Reproduced with permission of Oxford University Press and Richard W. Price. Requires documented HIV-1 infection and exclusion of other causes of acquired cerebral dysfunction, such as opportunistic infections and metabolic encephalopathies. In 1991, the AIDS Task Force of the American Academy of Neurology (AAN) defined the criteria for HAD and a milder condition, minor cognitive motor disorder (MCMD) [12]. The advent of cART with at least three antiretroviral drugs, in the mid-1990s, dramatically improved the outlook for HIV+ patients with access to treatment. Death rates fell, as did the incidence of HAD along with neurologic and systemic OIs [13, 14]. With improved survival, the prevalence of HAD increased, as did the frequency of less severe degrees of cognitive impairment [9–11, 13]. Progression over months was less common in treated patients, suggesting that cART had beneficial effects on the natural history of HAD, including slowing down the course. Dementia is now seen at higher CD4 counts than in the pre-cART era [13]. Risk factors for cognitive impairment in patients on cART include advancing age, particularly older than 50 years of age, and possibly hepatitis C coinfection [10]. With increasing numbers of older HIV+ patients, due in part to long-term survival on cART, distinguishing HAD and related conditions from age-related neurodegenerative disorders could become a more common clinical problem [15]. In 2007, a working group of the National Institute of Mental Health and the National Institute of Neurological Disorders and Stroke revised the AAN research nosology and associated diagnostic criteria, in part reflecting changes in clinical features and epidemiology in the era of cART [16]. This most recent update defined three conditions as comprising the HIV-associated neurocognitive disorders (HANDs): asymptomatic neurocognitive impairment, mild neurocognitive disorder, and HIV-1-associated dementia. The revised criteria emphasize acquired cognitive impairment, assessed by neuropsychological testing when possible, in at least two cognitive domains, typically impaired attention and learning with slowed information processing. The updated HAD criteria usually correspond to MSK stage score of two or greater. Significant functional impairment and the absence of delirium or other evident causes for dementia (including cerebral infection or neoplasm, stroke, other neurologic diseases, or substance abuse) are other key elements. The prevalence of HAND is approximately 50%, with a shift from HAD to milder forms in the era of cART [10, 16]. The differential diagnosis of cognitive impairment in patients with known HIV infection is broad. Substance abuse, psychiatric disorders, toxic-metabolic encephalopathies, neurosyphilis (discussed later in this chapter), and neurodegenerative disorders are considerations throughout the course of HIV disease. At CD4 counts below 200 cells/mm3, cerebral malignancies and OIs are important and life-threatening diagnostic concerns [8–10]. Depression is common in HIV+ patients and can be difficult to distinguish from early HAD. Primary central nervous system (CNS) lymphoma in the setting of HIV classically causes altered mental status with focal cerebral dysfunction or headache, typically evolving over months. The predilection of this B-cell lymphoma for periventricular regions and the corpus callosum [9] means that patients with bifrontal involvement may present primarily with personality change and cognitive impairment with minimal visual or motor dysfunction, at least in the early stages. Progressive multifocal leukoencephalopathy, an infection of oligodendrocytes by JC virus, typically manifests as worsening lateralized visual or motor impairment, evolving over months, consistent with asymmetric hemispheric white matter involvement, or occasionally as progressive brainstem dysfunction, but presentation with dementia has been reported [17]. Cerebral toxoplasmosis is a common cerebral OI in the setting of HIV/AIDS. Most commonly, this protozoal infection presents with headache, fever, focal cerebral dysfunction, impaired alertness, and other features of expanding cerebral mass lesions, although rarer meningoencephalitic forms with unusually bland neuroimaging have resembled HAD [18]. Cryptococcal meningitis, another common cerebral OI in HIV+ patients, is discussed later in this chapter. Cytomegalovirus (CMV) encephalitis sometimes presents as a rapidly progressive dementia, usually in very advanced AIDS, when CD4 count falls below 50 cells/mm3 [9, 19, 20]. Nearly one-third of the patients with CMV encephalitis and concurrent HIV infection have evidence of brainstem or cerebellar dysfunction [20]. Concomitant myelitis, radiculitis, retinitis, esophagitis, hepatitis, or colitis can also be useful diagnostic clues, since these are other common sites of CMV infection [9, 20]. CSF findings of polymorphonuclear pleocytosis with hypoglycorrhachia, resembling bacterial meningitis, and neuroimaging revealing ventriculitis are also suggestive, although the absence of either or both does not exclude CMV encephalitis. CSF CMV polymerase chain reaction (PCR) is more specific than sensitive for the diagnosis. CMV encephalitis is fatal without treatment but, if caught early, might respond to ganciclovir or foscarnet [9, 20]. It should be emphasized that throughout the HIV/AIDS epidemic, dementia has been reported as the presenting manifestation of previously undiagnosed HIV infection [21, 22]. HIV testing is thus an appropriate part of the evaluation for patients with unexplained cognitive impairment. HIV entry into the CNS has been demonstrated early in the course of infection, via infected monocytes [9–11, 23]. Occasionally, this is associated with typical aseptic meningitis but more commonly with asymptomatic mild lymphocytic CSF pleocytosis and elevated protein [9–11, 23–25]. Productive infection of neurons has not been demonstrated, suggesting that indirect mechanisms of brain injury, such as chronic inflammation or viral protein neurotoxicity, may underlie the myelin pallor and multinucleated giant cells seen in patients dying with HAD in the pre-cART era [5, 10, 23]. As the epidemiology and natural history of HANDs in the cART era have evolved, so also has the neuropathology, with more prominent inflammation in the brains of treated patients [10]. The diagnostic evaluation for HIV+ patients with cognitive dysfunction should include review of medications and screening for depression and substance use disorders, along with appropriate blood tests for thyroid disease, vitamin B12 deficiency, and neurosyphilis. Neuroimaging, with and without contrast when safe and feasible, to exclude multifocal OIs and malignancies typically reveals cerebral atrophy [9, 10]. MRI is superior to CT (Figure 13.1) in demonstrating symmetric, ill-defined, nonenhancing white matter hyperintensities on T2-weighted images [10], but these are neither necessary nor sufficient for a diagnosis of HAD. CSF protein often is mildly elevated; white cell count depends in part on CD4+ count [24, 25]. Pleocytosis is unusual among patients with CD4 counts below 50 cells/mm3, regardless of treatment status [24]. At higher CD4+ counts, patients not on cART, with or without neurologic symptoms, may demonstrate mildly elevated CSF white count (<30 cells/mm3). Pleocytosis normalizes within months of virologic control of HIV on cART, in concert with CSF HIV viral load falling near or below the limit of detection [11, 24, 25]. Small clinical studies in the pre-cART era demonstrated that zidovudine monotherapy, albeit at doses higher than typically used currently, slowed the progression of dementia [9, 23]. Zidovudine, unlike most other antiretrovirals in use at the time, achieved good CSF levels, generating debate about the degree to which CSF (and, it is assumed, cerebral) penetration is important in treating HAD [9, 10, 23, 26]. In addition to zidovudine, nevirapine and ritonavir-boosted indinavir penetrate well into the CNS, followed by abacavir, emtricitabine, delavirdine, efavirenz, and indinavir; boosted darunavir, fosamprenavir, or lopinavir; and maraviroc and raltegravir [10, 26]. Other antiretroviral drugs do not penetrate the CNS as well [9–11]. Patients who develop HANDs while not on cART should be started on therapy [9–11, 23]; whether patients who develop HANDs while on cART benefit from modifying the regimen toward drugs with good CNS penetration remains uncertain. Controlled trials of adjunctive antioxidant and neuroprotective agents have been disappointing [9, 10]. The patient was begun on abacavir, lamivudine, zidovudine, and nevirapine, which led to an increased CD4+ T-cell count and a decrease in viral load to undetectable levels. Over the next year, his attention, memory, and gait improved. Subsequently, he suffered a relapse of his methamphetamine use, nonadherence with his antiretroviral regimen, and was lost to follow-up. Subacute sclerosing panencephalitis (SSPE) is caused by cerebral infection with defective measles virus occurring long after acute measles infection [27–32]. Incidence estimates range from four to nearly 30 SSPE cases per 100 000 measles cases [27]. As a consequence of a preventable childhood infection, SSPE has become quite rare in countries with effective measles vaccination programs, and the diagnosis is easily missed [27, 28]. SSPE is typically a disease of childhood and adolescence, although adult-onset cases have been reported, with symptoms beginning as late as 49 years of age [28, 29]. Acute measles acquired very early in life increases the risk for SSPE, and pregnancy may also be a risk factor [27–29]. Symptoms begin months to decades after acute measles, which might not have been diagnosed, and include cognitive impairment, behavioral and personality changes, visual dysfunction, myoclonus, and seizures, usually developing over months [27, 28]. The myoclonus is typically generalized, without associated loss of consciousness and is common, but not always present [27, 28]. When myoclonus is present, the differential diagnosis includes prion disorders, corticobasal syndrome, dementia with Lewy bodies, hypoxia, thyrotoxic encephalopathy, and progressive myoclonic epilepsies such as mitochondrial cytopathies, Lafora’s disease, neuronal ceroid lipofuscinoses, Unverricht–Lundborg syndrome, and the sialidoses [27]. In the absence of myoclonus, the clinical syndrome suggests a neurodegenerative disorder, making the recognition of SSPE especially challenging [30]. The diagnosis should be considered through middle age in patients with cognitive or behavioral decline, particularly associated with myoclonus. Neuroimaging may be normal in early SSPE. As the disease advances, subcortical and periventricular white matter and basal ganglia abnormalities may be seen, progressing to hemispheric, brainstem, and cerebellar atrophy [27]. EEG may be normal or nonspecifically slow in early disease but typically demonstrates synchronous, stereotyped high-voltage periodic complexes usually associated with myoclonic jerks in midstage SSPE [27]. Routine CSF studies may be entirely normal or show mild lymphocytic pleocytosis and protein elevation, with elevated CSF gamma globulin concentration [27, 28, 30]. Antimeasles antibodies are elevated in CSF and establish the diagnosis, in the appropriate clinical setting and with characteristic EEG abnormalities [27–31]. Brain biopsy might be necessary in rare cases, such as in very early or advanced disease, when EEG may not show periodic complexes [27]. Neuropathologic findings include parenchymal inflammation in hemispheres spreading to brainstem and spinal cord, with demyelination and neuronal and glial viral inclusions [27, 32]. The occipital lobes are often involved in early disease, perhaps accounting for visual symptoms, although retinitis or optic nerve involvement also occurs [27, 32]. Fulminant cases resembling acute viral encephalitis have been reported [29], but the typical course is progression to death over several years [27, 28]. Oral isoprinosine alone or in combination with alpha interferon (intramuscular, intravenous, or intrathecal) or ribavirin (intraventricular or intravenous) might prolong survival, with varying degrees of symptomatic improvement [27]. Myoclonus is managed with benzodiazepines and antiepileptic agents but can be refractory to treatment [27]. Previous concerns that the measles vaccine might cause SSPE have not been realized. Prevention with vaccination remains the best intervention for this devastating infection [27]. Seroprevalence rates for antibodies to hepatitis C virus (HCV), indicating exposure to the hepatotropic single-stranded RNA virus, are below 2% in most industrialized nations and higher in Africa (0.8% in Ethiopia to more than 15% in Rwanda and Egypt), Latin America (0.7% in Mexico to 11.2% in Bolivia), and parts of Asia (5.6% in Thailand, 6.2% in Vietnam, 10.7% in Mongolia) [33]. Most infected individuals develop chronic liver disease, which can progress to cirrhosis and hepatocellular carcinoma [33–35]. Extrahepatic disease is common, and neurologic complications include peripheral neuropathy and stroke, in addition to hepatic encephalopathy from portal hypertension [35]. Coinfection with HIV and HCV is common, as both viruses are transmissible by exposure to blood, including injection drug use [33]. Even in the absence of cirrhosis, fatigue and functionally limiting memory impairment are common complaints in early HCV disease [35–37]. Abnormal neuropsychological testing, particularly in attention and executive function, compared to controls has been reported in patients with HCV without cirrhosis in many, but not all, studies [36]. Proton magnetic resonance spectroscopy demonstrated elevated choline to creatine ratio in the basal ganglia of patients with HCV and histologically mild liver disease [37]. Selection of proper testing batteries, control for psychiatric comorbidities, and relevance of abnormal functional imaging are among many methodologic concerns in these studies [35, 36]. In the setting of HIV coinfection, HCV infection was associated with adverse effects on cognitive function in patients with advanced HIV disease [38], but not in the setting of well-controlled HIV infection [39] or hemophilia [40]. Interestingly, HCV is not strictly hepatotropic and can replicate in peripheral blood mononuclear cells [33, 41]. The identification in the brain of macrophages and microglia harboring HCV is reminiscent of the pathogenesis of HAD and related disorders [10, 41]. Treatment for HCV consists of ribavirin with alpha interferon, the latter of which can cause depression or cognitive impairment [36]. Viruses are common causes of acute infectious encephalitis, although extensive diagnostic evaluation often does not reveal the specific pathogen [42]. Herpesviruses, neurotropic double-stranded DNA viruses, are important diagnostic considerations year-round. Herpes simplex virus 1 (HSV-1) accounts for 5–10% of encephalitis cases in the United States [42–44]. Reactivation of latent HSV-1 in trigeminal ganglia most commonly causes labial vesicular lesions known as cold sores and, rarely, spreads proximally to the brain, resulting in HSV-1 encephalitis [44, 45]. In addition to fever, headache, and seizures, common presenting symptoms include behavioral and personality change and aphasia, consistent with involvement of one or both medial temporal lobes or orbitofrontal cortices [44, 45]. High-dose intravenous acyclovir lowered mortality to 28%, compared to 70% in historical controls [46]. Even with treatment, many survivors have persistent neuropsychological impairment including anterograde and retrograde amnesia, anomia, semantic memory deficit, executive dysfunction, mood disorders, and dementia [43–46]. Arbovirus (arthropod-borne virus) infections occur classically as summer outbreaks and are important causes of encephalitis globally. West Nile virus, first isolated from an African patient in 1937, rapidly became endemic in North America after a 1999 outbreak in New York City [47]. The mosquito-borne virus most frequently causes the acute febrile illness known as West Nile fever and, occasionally, West Nile neuroinvasive disease involving the meninges, brain, or anterior horn cells [47, 48]. Impaired function or quality of life was noted 18 months after the acute illness in half or more patients after West Nile infection, even those without neuroinvasive disease [48]. Neuropsychological testing revealed psychomotor slowing [47]. Acute mortality for tick-borne encephalitis, the predominant European arbovirus, is low, but persistently impaired memory is a common long-term complication, with normal cognitive testing in only 10% of survivors and dementia evident in one-third [49]. Data are even more scant for neuropsychological outcomes from other forms of viral encephalitis [45]. Cognitive dysfunction tends to remain stable or improve slowly over time [50]. A middle-aged man was brought to the emergency department for cognitive decline and personality change. His family described him as progressively disoriented, hostile, and moody for over several months. Earlier that week, he had struck his wife. The patient did not have any pain, incontinence, or a gait disorder. He took atenolol for hypertension and had no other medical problems and no previous psychiatric history. The patient worked as a kitchen assistant; did not use tobacco, alcohol, or illicit drugs; and had no family history of dementia. On examination, he was disheveled, mildly agitated, and afebrile, with normal vital signs and no meningismus. The neurologic exam was limited by the rare dialect that he spoke—he was oriented only to name, with normal speech and language, and impaired registration. Other exam findings included normal pupils, slightly brisk right patellar and bilateral ankle reflexes, flexor plantar responses, and normal proprioception and gait. Electrolytes and liver and renal function tests were normal. Head CT showed marked generalized atrophy for his age, without hydrocephalus or mass lesion, with no additional abnormalities on MRI (Figure 13.2). Lumbar puncture showed normal opening pressure of 80 mm H2O. CSF protein was elevated at 110 mg/dL and glucose was slightly low at 42 mg/dL (serum 82 mg/dL). Tube #1 of CSF showed 2500 RBC/mm3 and 19 WBC/mm3 (84% lymphocytes). Tube #4 showed 695 RBC/mm3 and 12 WBC/mm3 (84% lymphocytes). VDRL was 1:128 in serum and VDRL was 1:16 in CSF, confirming a diagnosis of neurosyphilis. Thyroid-stimulating hormone and vitamin B12 were normal, and HIV serology was negative. An EEG showed diffuse slowing. The patient underwent treatment for neurosyphilis with penicillin four million units intravenously every 4 h for 2 weeks. Behavioral symptoms were managed with risperidone, haloperidol, and lorazepam (case continued below). Figure 13.2 Neuroimaging in general paresis noncontrast head CT (a), postgadolinium T1-weighted (b), and proton density (c) MRI images obtained from a middle-aged patient with general paresis (see text) show severe cerebral atrophy for age. The incidence of primary syphilis in the United States has risen in recent years, especially among men who have sex with men [51–54]. Caused by infection with the bacterium Treponema pallidum spp. pallidum (hereafter referred to as T. pallidum
Infectious causes of dementia
Introduction
Viruses
Human immunodeficiency virus (HIV)-associated dementia
Stage 0
Normal
Stage 0.5
Absent, minimal, or equivocal symptoms of cognitive or motor dysfunction characteristic of ADC
Mild signs (snout reflex, slowed ocular or limb movements) without impairment of work or capacity to perform activities of daily living (ADL)
Normal gait and strength
Stage 1
Able to perform all but the more demanding aspects of work or ADL
Unequivocal evidence (symptoms, signs, neuropsychological testing) of functional intellectual or motor impairment
Can walk without assistance
Stage 2
Cannot work or maintain more demanding aspects of daily life but able to perform basic self-care activities
Ambulatory but may require single prop
Stage 3
Major intellectual incapacity (unable to follow news or personal events, sustain complex conversation; considerable slowing of all output) or motor disability (unable to walk unassisted, requiring walker or personal support, usually with slowing and clumsiness in arms)
Stage 4
Nearly vegetative, with rudimentary intellectual and social comprehension and responses; nearly or absolutely mute
Paraparetic or paraplegic with double incontinence
Case follow-up
Subacute sclerosing panencephalitis
Hepatitis C
Cognitive sequelae of viral encephalitis
Bacteria
Neurosyphilis: General paresis
Case
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel







Full access? Get Clinical Tree


