CHAPTER 43 Antidepressants
OVERVIEW
A large number of compounds have been developed to treat depression. Traditionally, these compounds have been called “antidepressants,” even though most of these drugs are also effective in the treatment of a number of anxiety disorders (such as panic and obsessive-compulsive disorder [OCD]) and a variety of other conditions (Table 43-1). The precursors of two of the major contemporary antidepressant families, the monoamine oxidase inhibitors (MAOIs) and the tricyclic antidepressants (TCAs), were discovered by serendipity in the 1950s.
Table 43-1 Indications for Antidepressants
| Major depressive disorder and other unipolar depressive disorders Bipolar depression Panic disorder Social anxiety disorder Generalized anxiety disorder Posttraumatic stress disorder Obsessive-compulsive disorder (e.g., clomipramine and SSRIs) Depression with psychotic features (in combination with an antipsychotic drug) Bulimia nervosa Neuropathic pain (tricyclic drugs and SNRIs) Insomnia (e.g., trazodone, amitriptyline) Enuresis (imipramine best studied) Atypical depression (e.g., monoamine oxidase inhibitors) Attention-deficit/hyperactivity disorder (e.g., desipramine, bupropion) |
SNRIs, Serotonin norepinephrine reuptake inhibitors; SSRIs, selective serotonin reuptake inhibitors.
Specifically, the administration of iproniazid, an antimycobacterial agent, was first noted to possess antidepressant effects in depressed patients suffering from tuberculosis.1 Shortly thereafter, iproniazid was found to inhibit MAO, involved in the catabolism of serotonin, norepinephrine, and dopamine.
In parallel, imipramine was initially developed as an antihistamine, but Kuhn2 discovered that of some 500 imipramine-treated patients with various psychiatric disorders, only those with endogenous depression with mental and motor retardation showed a remarkable improvement during 1 to 6 weeks of daily imipramine therapy. The same compound was also found to inhibit the reuptake of serotonin and norepinephrine.3,4 Thus, it was the discovery of the antidepressant effects of iproniazid and imipramine that led to the development of the MAOIs and TCAs, but also such discovery was instrumental in the formulation of the monoamine theory of depression. In turn, guided by this theory, the subsequent development of compounds selective for the reuptake of either serotonin or norepinephrine or both was designed, rather than accidental. As a result, over the last few decades, chemical alterations of these first antidepressants have resulted in the creation of a wide variety of monoamine-based antidepressants with a variety of mechanisms of action. The antidepressant drugs are a heterogeneous group of compounds that have been traditionally subdivided into major groups according to their chemical structure or, more commonly, according to their effects on monoamine neurotransmitter systems: selective serotonin reuptake inhibitors (SSRIs); TCAs and the related cyclic antidepressants (i.e., amoxapine and maprotiline); MAOIs; serotonin norepinephrine reuptake inhibitors (SNRIs); norepinephrine reuptake inhibitors (NRIs); norepinephrine/dopamine reuptake inhibitors (NDRIs); serotonin receptor antagonists/agonists; and alpha2-adrenergic receptor antagonists. Because they overlap, the mechanisms of action and the indications for use for the antidepressants are discussed together, but separate sections are provided for their method of administration and their side effects.
MECHANISM OF ACTION
The precise mechanisms by which the antidepressant drugs exert their therapeutic effects remain unknown, although much is known about their immediate actions within the nervous system. All of the currently marketed antidepressants interact with the monoamine neurotransmitter systems in the brain, particularly the norepinephrine and serotonin systems, and to a lesser extent the dopamine system. Essentially all currently marketed antidepressants have as their molecular targets components of monoamine synapses, including the reuptake transporters (that terminate the action of norepinephrine, serotonin, or dopamine in synapses), monoamine receptors, or enzymes that serve to metabolize monoamines. What remains unknown is how these initial interactions produce a therapeutic response.5 The search for the molecular events that convert altered monoamine neurotransmitter function into the lifting of depressive symptoms is currently a matter of very active research.
Since TCAs and MAOIs were the first antidepressants to be discovered and introduced, this was initially interpreted as suggesting that antidepressants work by increasing noradrenergic or serotonergic neurotransmission, thus compensating for a postulated state of relative monoamine “deficiency.” However, this simple theory could not fully explain the action of antidepressant drugs for a number of reasons. The most important of these include the lack of convincing evidence that depression is characterized by a state of inadequate or “deficient” monoamine neurotransmission. In fact, the results of studies testing the monoamine depletion hypothesis in depression have yielded inconsistent results.5 Moreover, blockade of monoamine reuptake or inhibition of monoamine degradation occurs rapidly (within hours) following monoamine reuptake inhibitor or MAOI administration, respectively. However, treatment with antidepressants for less than 2 weeks is unlikely to result in a lifting of depression; it has been consistently observed and reported that remission of depression often requires 4 weeks of treatment or more. These considerations have led to the idea that inhibition of monoamine reuptake or inhibition of MAO by antidepressants represents an initiating event. The actual therapeutic actions of antidepressants, however, result from slower adaptive responses within neurons to these initial biochemical perturbations (“downstream events”).6 Although research geared toward understanding the therapeutic actions of antidepressants has been challenging, receptor studies have been useful in understanding and predicting some of the side effects of contemporary antidepressants. For example, the binding affinity of antidepressants at muscarinic cholinergic receptors generally parallels the prevalence of certain side effects during treatment (e.g., dry mouth, sedation, weight gain, constipation, urinary hesitancy, poor concentration). Similarly, treatment with agents that have high affinities for histamine H1 receptors (e.g., doxepin and amitriptyline) appears to be more likely to result in sedation, dry mouth, and increased appetite. Such information is very useful to clinicians and patients when making treatment decisions or to researchers when attempting to develop new antidepressants.
The architecture of the monoamine neurotransmitter systems in the central nervous system (CNS) is based on the synthesis of the neurotransmitter within a restricted number of nuclei within the brainstem, with neurons projecting widely throughout the brain and, for norepinephrine and serotonin, the spinal cord as well.5 Norepinephrine is synthesized within a series of nuclei in the medulla and pons, of which the largest is the nucleus locus coeruleus. Serotonin is synthesized in the brainstem raphe nuclei. Dopamine is synthesized in the substantia nigra and the ventral tegmental area of the midbrain. Through extensive projection networks, these neurotransmitters influence a large number of target neurons in the cerebral cortex, basal forebrain, striatum, limbic system, and brainstem where they interact with multiple receptor types to regulate arousal, vigilance, attention, sensory processing, emotion, and cognition (including memory).7
Norepinephrine, serotonin, and dopamine are removed from synapses after release by reuptake, mostly into presynaptic neurons.5 This mechanism of terminating neurotransmitter action is mediated by specific norepinephrine, serotonin, and dopamine reuptake transporter proteins. After reuptake, norepinephrine, serotonin, and dopamine are either reloaded into vesicles for subsequent release or broken down by MAO. MAO is present in two forms (MAOA and MAOB), which differ in their substrate preferences, inhibitor specificities, tissue expression, and cell distribution. MAOA preferentially oxidizes serotonin and is irreversibly inactivated by low concentrations of the acetylenic inhibitor clorgyline. MAOB preferentially oxidizes phenylethylamine (PEA) and benzylamine and is irreversibly inactivated by low concentrations of pargyline and deprenyl.5 Dopamine, tyramine, and tryptamine are substrates for both forms of MAO. Catecholamines are also broken down by catechol O-methyltransferase (COMT), an enzyme that acts extracellularly.
The classification of antidepressant drugs has perhaps focused too narrowly on synaptic pharmacology (i.e., “immediate effects”), and has certainly failed to take into account molecular and cellular changes in neural function that are brought about by the chronic administration of these agents.5 For example, it has been postulated that changes in post-receptor signal transduction may account for the aforementioned characteristic lag-time between the time a drug is administered and the drug-induced resolution of a depressive episode. In fact, a recent study has shown hippocampal neurogenesis to occur following chronic antidepressant treatment in animal models.8 In parallel, rapid activation of the TrkB neurotrophin receptor and PLC gamma-1 signaling has been described with almost all antidepressant drugs, a possible mechanism by which the process of neuronal neurogenesis observed following chronic administration of antidepressants may occur.9 Alternatively, one could postulate that this lag phase in antidepressant action may be related to a reorganization of neuronal networks, postulated as a potential “final common pathway” for antidepressant effects to occur.5 Nevertheless, further research is urgently needed in order to help us better understand the specific effects of the antidepressants and what constitutes illness and recovery in depression.
Mechanism of Action of Selective Serotonin Reuptake Inhibitors
At therapeutically relevant doses, the SSRIs exhibit significant effects primarily on serotonin reuptake in the human brain.10 Some SSRIs also appear to have effects on other monoamine transporters, with sertraline demonstrating modest dopamine reuptake inhibition, and paroxetine and fluoxetine demonstrating modest norepinephrine reuptake inhibition.10 In addition, fluoxetine, particularly the R-isomer, has mild 5-HT2A and 5-HT2C antagonist activity. Non-monoaminergic effects have also been described for some of the SSRIs, including moderate and selective effects on glutamate receptor expression and editing.11 The SSRIs have minimal or no affinity for muscarinic cholinergic, histaminergic, and adrenergic receptors, with the exception of paroxetine (which is a weak cholinergic receptor antagonist), citalopram (which is a weak antagonist of the histamine H1 receptor), and sertraline (which has weak affinity for the alpha1 receptors).10 Overall, the affinity of these agents for these specific receptors is lower than those of the TCAs, resulting in a milder side-effect profile. Similarly, the lack of significant action for the remaining SSRIs on these receptors is also thought to contribute to the milder side-effect profile of these agents compared with the TCAs.
Mechanism of Action of Serotonin Norepinephrine Reuptake Inhibitors
Unlike the TCAs, SNRIs inhibit the reuptake of serotonin more potently than the reuptake of norepinephrine.10 Similar to most SSRIs, the SNRIs have minimal or no affinity for muscarinic cholinergic, histaminergic, and adrenergic receptors.10 Interestingly enough, administration of these drugs has been shown to prevent a decrease in cell proliferation and BDNF expression in rat hippocampus observed with chronic stress, a study that offers further insights into the “downstream effects” of these agents.12 In parallel, recent data suggest that chronic treatment with the SNRI duloxetine not only produces a marked up-regulation of BDNF mRNA and protein, but may also affect the subcellular redistribution of neurotrophin, potentially improving synaptic plasticity.13
Mechanism of Action of Norepinephrine Reuptake Inhibitors
At therapeutically relevant doses, the NRIs have significant effects primarily on norepinephrine reuptake, although the NRI atomoxetine is also a weak inhibitor of serotonin uptake.14 NRIs also appear to have several nonmonoaminergic properties. Specifically, the NRI reboxetine also appears to functionally inhibit nicotinic acetylcholine receptors.15 In addition, in rats, atomoxetine has been shown to increase in vivo extracellular levels of acetylcholine (Ach) in cortical but not subcortical brain regions, with a mechanism dependent on norepinephrine alpha1 and/or dopamine D1 receptor activation.16 Furthermore, the major human metabolite of atomoxetine (4-hydroxyatomoxetine) is a partial agonist of the kappa-opioid receptor.17 Finally, reboxetine has also been found to be the antidepressant that affects glutamate receptors (GluR) most, with a decrease of GluR3 expression.11
Mechanism of Action of Serotonin Receptor Agonist/Antagonists
Both trazodone (Desyrel) and nefazodone (Serzone) are relatively weak inhibitors of serotonin and norepinephrine uptake, and they primarily block serotonin 5-HT2A receptors (in some cases, demonstrating partial agonist properties as well).18–21 They also share a metabolite, m-chlorophenylpiperazine (mCPP), which acts as a serotonin 5-HT2C agonist and appears to be able to release serotonin presynaptically.22 Trazodone also appears to stimulate the mu1– and mu2-opioid receptors23 and is a potent agonist of the serotonin 5-HT2C receptors, which, when activated,24,25 may inhibit NMDA-induced cyclic GMP elevation. Since trazodone is also a weak inhibitor of serotonin reuptake as well, the overall effect of trazodone appears to be an increase in extracellular levels of serotonin in the brain.26 This effect explains the fact that trazodone treatment has been associated with the occurrence of a serotonin syndrome.27 Both trazodone and (although to a lesser degree) nefazodone are potent blockers of the alpha1-adrenergic receptor.
Agomelatine, a newer agent, is a selective 5-HT2C antagonist and also an agonist at various melatonergic receptors. The 5-HT2C antagonism properties of agomelatine have been thought to be responsible for increases in frontocortical dopaminergic and adrenergic activity in animals during administration of agomelatine.28
Buspirone and gepirone act as full agonists at serotonin 5-HT1A autoreceptors and are generally, but not exclusively, partial agonists at postsynaptic serotonin 5-HT1A receptors.29 Buspirone and gepirone show weak alpha1-adrenoceptor affinity but significant and selective alpha1-adrenoceptor intrinsic efficacy, which was expressed in a tissue- and species-dependent manner.30 They also show weak dopamine D2 antagonism properties.31 The latter effect is thought to lead to excitation of noradrenergic cell firing,31 antagonizing primarily presynaptic inhibitory dopamine D2 autoreceptors at dopaminergic neurons.32 Buspirone also has potent alpha2-adrenoceptor antagonist properties via its principal metabolite, 1-(2-pyrimidinyl)-piperazine.33,34
Mechanism of Action of Norepinephrine/Dopamine Reuptake Inhibitors
The NDRIs primarily block the reuptake of dopamine and norepinephrine and have minimal or no affinity for presynaptic or postsynaptic monoamine receptors. The mechanism of action of bupropion has not been fully elucidated, although it appears to primarily block the reuptake of both dopamine and norepinephrine.35 Bupropion and its metabolites have been shown to be able to inhibit striatal uptake of the selective dopamine transporter (DAT)-binding radioligand (11)C-bCIT-FE in vivo,36 and to have mild affinity for the norepinephrine transporter,37 although some researchers have argued that the effect of bupropion on norepinephrine is primarily through an increase in presynaptic norepinephrine release.38 Whatever the exact mechanism may be, it appears that the overall effect of bupropion is a dose-dependent increase in brain extracellular dopamine and norepinephrine concentrations.39 It also appears that bupropion acts as an antagonist at alpha3beta2 and alpha3beta4 nAChRs in rat striatum and hippocampus, respectively, across the same concentration range that inhibits DAT and norepinephrine transporter (NET) function.40
Mechanism of Action of Alpha2-Adrenergic Receptor Antagonists
The alpha2-adrenergic receptor antagonists (e.g., mirtazapine and mianserin) appear to enhance the release of both serotonin and norepinephrine by blocking auto- and hetero-alpha2 receptors.41 Since mirtazapine appears to be a blocker of serotonin 5-HT2 and 5-HT3 receptors as well, it is thought to enhance the release of norepinephrine and also enhance 5-HT1A–mediated serotonergic transmission.42 Mirtazapine was the first alpha2-adrenergic receptor antagonist to be approved by the Food and Drug Administration (FDA) for depression. Mirtazapine is also a potent histaminergic H1-receptor antagonist.10 Mianserin, also an alpha2-noradrenergic receptor antagonist and a serotonin 5-HT2 antagonist, is available in Europe (brand name: Lantanon) and is not FDA-approved.
Mechanism of Action of Tricyclic Antidepressants
TCAs, referred to as such because they share a chemical structure with two joined benzene rings, have been in use for almost half a century for the treatment of depression. With the exception of clomipramine, and in contrast with the SNRIs, the TCAs inhibit the reuptake of norepinephrine more potently than the reuptake of serotonin. Recent studies have also shown that five of the TCAs bind to the S1S2 domain of the GluR2 subunit of the AMPA receptor, suggesting an effect of TCAs on the glutamergic system.43 In addition, doxepin, amitriptyline, and nortriptyline also inhibit glycine uptake by blocking the glycine transporter 1b (GLYT1b) and glycine transporter 2a (GLYT2a) to a similar extent.44 Amoxapine displays a selective inhibition of GLYT2a behaving as a tenfold more efficient inhibitor of this isoform than of GLYT1b44 and is also a dopamine D2 receptor antagonist.45 Interestingly, in vitro data suggest that trimipramine and clomipramine have comparable affinity for the dopamine D2 receptor.46 The TCAs, to varying degrees, are also fairly potent blockers of histamine H1 receptors, serotonin 5-HT2 receptors, muscarinic acetylcholine receptors, and alpha1-adrenergic receptors.10,46
Mechanism of Action of Monoamine Oxidase Inhibitors
MAOIs act by inhibiting MAO, an enzyme found on the outer membrane of mitochondria, where it catabolizes (degrades) a number of monoamines including dopamine, norepinephrine, and serotonin. Specifically, following their reuptake from the synapse into the presynaptic neuron, norepinephrine, serotonin, and dopamine are either loaded into vesicles for subsequent re-release or broken down by MAO. MAO is present in two forms (MAOA and MAOB), which differ in their substrate preferences, inhibitor specificities, tissue expression, and cell distribution. MAOA preferentially oxidizes serotonin and is irreversibly inactivated by low concentrations of the acetylenic inhibitor clorgyline. MAOB preferentially oxidizes phenylethylamine (PEA) and benzylamine and is irreversibly inactivated by low concentrations of pargyline and deprenyl. Dopamine and the dietary (exogenous) amines, tyramine and tryptamine, are substrates for both forms of MAO.47 In the gastrointestinal (GI) tract and the liver, MAO catabolizes a number of dietary pressor amines (such as dopamine, tyramine, tryptamine, and phenylethylamine).48 For this reason, consumption of certain foods (that contain high levels of dietary amines) while on an MAOI may precipitate a hypertensive crisis, characterized by hypertension, hyperpyrexia, tachycardia, tremulousness, and cardiac arrhythmias.49 The same reaction may also occur during co-administration of dopaminergic agents and MAOIs, while the co-administration of MAOIs with other antidepressants that potentiate serotonin could result in serotonin syndromes due to toxic CNS serotonin levels. The serotonin syndrome is characterized by alterations in cognition (e.g., disorientation and confusion), behavior (e.g., agitation and restlessness), autonomic nervous system function (e.g., fever, shivering, diaphoresis, and diarrhea), and neuromuscular activity (e.g., ataxia, hyperreflexia, and myoclonus).50–52 Since MAO enzymatic activity requires approximately 14 days to be fully restored, such food or medications should be avoided for 2 weeks following the discontinuation of an irreversible MAOI (“MAOI washout period”). Serotonergic and dopaminergic antidepressants are typically discontinued 2 weeks before the initiation of an MAOI, with the exception of fluoxetine, which needs to be discontinued 5 weeks in advance because of its much longer half-life.
Older MAOIs, including phenelzine (Nardil), tranylcypromine (Parnate), and isocarboxazid (Marplan), irreversibly inhibit the enzymatic activity of both MAOA and MAOB, while newer ones are relatively selective (brofaromine [Consonar] and moclobemide [Manerix] preferentially inhibit MAOA; selegiline [Eldepryl] selectively inhibits MAOB). In addition, while older MAOIs result in irreversible inhibition of MAO, some newer ones (such as moclobemide and brofaromine) result in reversible inhibition. Reversible MAOA-selective inhibitors are designed to minimize the risk of hypertensive crises, and patients on conventional doses of moclobemide do not need to strictly adhere to the low-tyramine diet, although, at very high doses (e.g., 900 mg/day of moclobemide), inhibition of MAOB also occurs.53 All MAOIs available in the United States are irreversible inhibitors of MAOA and MAOB activity. Newer MAOIs (such as brofaromine and moclobemide) reversibly inhibit MAOA activity.
More recently, additional pharmacological properties for the MAOIs have been revealed. MAOIs, for instance, also appear to inhibit the binding of [3H] quinpirole, a dopamine agonist with high affinity for D2 and D3 dopamine receptors.54,55 To complicate the pharmacology of MAOIs further, two of the MAOIs, selegiline and tranylcypromine, have methamphetamine and amphetamine as metabolites.56,57 In addition, phenelzine also elevates brain gamma-aminobutyric acid (GABA) levels, and as yet unidentified metabolites of phenelzine may be responsible for this effect.57 R(−)- but not S(+)-selegiline also appears to induce dopamine release by directly modulating ATP-sensitive potassium channels.58 Finally, (+)-tranylcypromine (TCP) has been shown to be more potent than (-)-TCP as an inhibitor of 5-HT uptake, whereas (-)-TCP has been shown to be more potent than (+)-TCP as an inhibitor of dopamine and norepinephrine uptake.59
Although the risk for serotonin syndromes may be lower than with the older MAOIs, and a number of studies suggested the safety of combining moclobemide with SSRIs,60–62 there have been a number of nonfatal63,64 and fatal serotonin syndromes involving the co-administration of moclobemide and SSRIs.65–70 For these reasons, the concomitant use of moclobemide and serotonergic agents should be avoided. In addition, the co-ingestion of moclobemide and SSRIs in overdose may result in death, which needs to be taken into account for patients at risk for suicide.66
CLINICAL USES OF ANTIDEPRESSANTS
Continuation and Maintenance Antidepressant Treatment
Originally, based on studies with TCAs, patients with unipolar depressive disorders were observed to be at high risk for relapse when treatment was discontinued within the first 16 weeks of therapy. Therefore, in treatment-responders, most experts favor a continuation of antidepressant therapy for a minimum of 6 months following the achievement of remission. The value of continuation therapy for several months to prevent relapse into the original episode has also been established for virtually all of the newer agents.71 Risk of recurrence after this 6- to 8-month continuation period (i.e., the development of a new episode after recovery from the index episode) is particularly elevated in patients with a chronic course before recovery, residual symptoms, and multiple prior episodes (three or more).72 For these individuals, the optimal duration of maintenance treatment is unknown but is assumed to be much longer in duration (measured in years). In fact, based on research to date, prophylactic efficacy of an antidepressant has been observed for as long as 5 years with clear benefit.73 In contrast to the initial expectation that maintenance therapy would be effective at dosages lower than that required for acute treatment, the current consensus is that full-dose therapy is required for effective prophylaxis.74 About 20% to 30% of patients who are treated with each of the classes of antidepressants will experience a return of depressive symptoms despite continued treatment. In such patients, a dose increase of the antidepressant is typically the first-line approach.75
Suicide Risk
Unlike the SSRIs and other newer agents, the MAOIs, TCAs, and related cyclic antidepressants (maprotiline and amoxapine) are potentially lethal in overdose. Thus, a careful evaluation of impulsiveness and suicide risk influences not only the decision as to the need for hospitalizing a person with depression, but also the choice of an antidepressant. For potentially suicidal or highly impulsive patients, the SSRIs and the other newer agents would be a better initial choice than a cyclic compound or an MAOI. Patients at elevated suicide risk who cannot tolerate these safer compounds or who do not respond to them should not receive large quantities or refillable prescriptions for TCAs or MAOIs. Generally, patients who are new to treatment or those at more than minimal risk for suicide or whose therapeutic relationship is unstable should receive a limited supply of any medication. Evaluation for suicide risk must continue even after the initiation of treatment. Although suicidal thoughts are often among the first symptoms to improve with antidepressant treatment, they may also be slow to respond to treatment, and patients may become demoralized before therapeutic efficacy is evident. Side effects (such as agitation and restlessness) and, most important, intercurrent life events may exacerbate suicidal thoughts before a full therapeutic response. Thus, rarely, for a variety of reasons, patients may temporarily become more suicidal following the initiation of treatment. Should such worsening occur, appropriate interventions may include management of side effects, more frequent monitoring, discontinuation of the initial treatment, or hospitalization. In 2004, the FDA asked manufacturers of almost all the newer antidepressant drugs to include in their labeling a warning statement that recommends close observation of adult and pediatric patients treated with these drugs for worsening depression or the emergence of suicidality. This warning was based on the analyses of clinical trials data that compared the relative risk of emergence of suicidal ideation on these drugs compared to placebo following initiation of treatment. The difference was small, but statistically significant. This finding underscores the need for good practice, which includes education of patients (and families if the patient is a child) about side effects of drugs (including the possible emergence of suicidal thoughts and behaviors), close monitoring (especially early in treatment), and the availability of a clinician in case suicidality emerges or worsens. A general consensus remains, however, that the risks associated with withholding antidepressant treatment from patients, including pediatric patients, with serious depression vastly outweighs the risks associated with the drugs by many orders of magnitude.
CHOICE OF AN ANTIDEPRESSANT
A large number of antidepressants are available (Table 43-2), including SSRIs, SNRIs, NRIs, NDRIs, serotonin receptor antagonists/agonists (nefazodone and trazodone), the alpha2-adrenergic receptor antagonists, TCAs and related compounds, and MAOIs. The available formulations and their typical dosages are listed in Table 43-2, and aspects of their successful use are listed in Table 43-3.
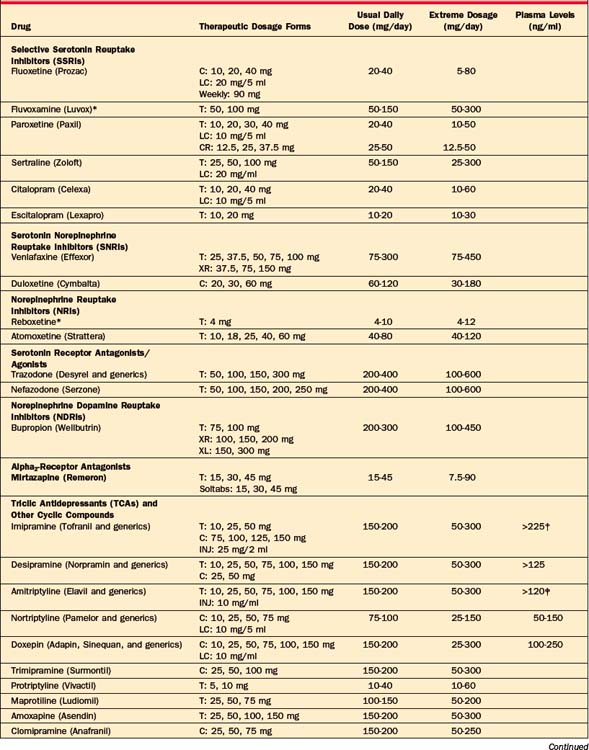
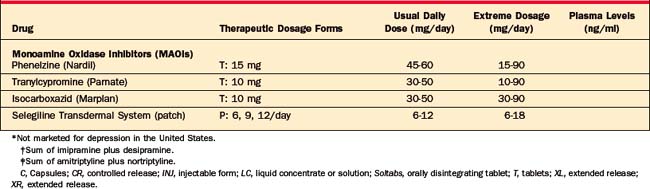
Table 43-3 Requirements for Successful Use of Antidepressants
NDRI, Norepinephrine/dopamine reuptake inhibitor; OCD, obsessive-compulsive disorder; SSRI, selective serotonin reuptake inhibitor.
Selective Serotonin Reuptake Inhibitors
The overall efficacy of the SSRIs in the treatment of depression is equivalent to that of older agents, including the TCAs and the MAOIs moclobemide76 and phenelzine,77 while all SSRIs appear to be equally effective in the treatment of depression.78 However, there is some evidence suggesting that that the SSRI escitalopram may be more effective than the remaining five SSRIs in the treatment of MDD,79 although this evidence remains controversial.
Because of their favorable side-effect profile, the SSRIs are used as first-line treatment in the overwhelming majority of cases, with more than 90% of clinicians in one survey indicating that SSRIs were their first-line treatment.80 Despite the tolerability and the widespread efficacy of the SSRIs, there is mounting evidence to suggest that depressed patients with certain characteristics (including co-morbid anxiety disorders81 and a greater number of somatic symptoms [such as pain, headaches, and fatigue]82) respond less well to SSRIs than those without such characteristics.
Dosage
Because of their relatively low side-effect burden, the starting dose of SSRIs is often the minimally effective daily dose: 10 mg for escitalopram (Lexapro); 20 mg for fluoxetine (Prozac), paroxetine (Paxil), and citalopram (Celexa); 50 mg for sertraline (Zoloft); 25 mg for paroxetine CR (Paxil CR); and 100 mg for fluvoxamine (Luvox). Starting at lower doses, and increasing the dose shortly thereafter (i.e., after 1 to 2 weeks) may further improve tolerability. Maximum therapeutic doses for SSRIs are typically onefold to fourfold greater than the starting dose. The dosages and formulations of the SSRIs marketed in the United States are listed in Table 43-2. Only one of the SSRIs, fluvoxamine, is not approved for the treatment of depression in the United States, as it is approved only for the treatment of OCD, although several placebo-controlled trials have demonstrated the efficacy of fluvoxamine in MDD.83,84
Side-Effect Profile
The most common side effects of the SSRIs are nausea, tremor, excessive sweating, flushing, headache, insomnia, activation or sedation, jitteriness, dizziness, rash, and dry mouth.85 Sedation does not appear to occur more often with any particular SSRI, while the SSRIs appear to be equally well tolerated and effective in the treatment of depressed patients, regardless of whether they cause insomnia, activation, or sedation.78 The use of SSRIs is also associated with the emergence of sexual dysfunction (including decreased libido, delayed ejaculation, impotence, and anorgasmia), or the worsening of preexisting sexual dysfunction in depression.86 These side effects tend to improve rapidly after temporary (“drug holiday”) discontinuation of the SSRIs, particularly those SSRIs with a shorter half-life,87 although prolonging such drug holidays carries a risk of withdrawal effects and of depressive relapse. Some patients treated with SSRIs may also experience cognitive symptoms (such as mental slowing and worsened attention), psychological symptoms (such as apathy and emotional blunting),88 and motor symptoms (such as bruxism and akathisia).89,90 Other, less common, adverse events associated with SSRI treatment include diarrhea, tremor, bruxism, rash, hyponatremia, hair loss, and the syndrome of inappropriate antidiuretic hormone (SIADH) secretion. There are also case reports of the SSRIs worsening motor symptoms in patients with Parkinson’s disease,91–93 as well as creating increased requirements for levodopa in Parkinson’s patients following initiation of an SSRI for depression.94 SSRIs have been associated with abnormal bleeding (e.g., bruising and epistaxis) in children and adults who have unremarkable routine hematological laboratory results except for abnormal bleeding time or platelet counts.95 A systematic study of this issue has failed to reveal abnormalities in platelet aggrega-tion, hematopoiesis, or coagulation profile in SSRI-treated patients.96 Although many patients may also experience reduced appetite and weight loss during the acute phase of treatment with SSRIs,97,98 any beneficial effects of the SSRIs with respect to weight loss do not seem to be sustained during the continuation and maintenance phases of treatment,97,98 while one study reveals a greater risk for significant weight gain during long-term treatment with paroxetine, but not fluoxetine or sertraline.99 Although SSRI-induced side effects appear to be well tolerated by most patients,100 for depressed patients who are unable to tolerate one SSRI, switching to another SSRI has been effective and well tolerated in most cases.101–104 For patients complaining of GI side effects with paroxetine, the continued-release formulation (Paxil CR), reported to have a lower incidence of nausea, may be used in place of the standard formulation.105 As with other antidepressants, the potential adverse neuroendocrine and skeletal effects of the SSRIs have yet to be systematically explored.106 However, a recent study from our group has shown that 4.5% of men and 22.2% of women with MDD developed new-onset hyperprolactinemia following SSRI treatment,107 and daily SSRI use in adults 50 years and older was associated with a twofold increased risk of fractures after adjustment for potential co-variates.108 The SSRIs also appear to possess the lowest toxicity in overdose.109 Finally, of all antidepressants, fluoxetine has the most extensive literature supporting its reproductive safety.110
SSRI Discontinuation Syndrome
A number of reports also describe discontinuation-emergent adverse events after abrupt cessation of SSRIs, including dizziness, insomnia, nervousness, irritability, nausea, and agitation.111,112 The risk of such adverse events occurring seems to be inversely related to the half-life of the SSRI, with fluoxetine reported as having a significantly lower risk than paroxetine in two studies.111,113 For more severe discontinuation-related adverse events, reinstitution of the SSRI and slow taper may be necessary to alleviate these symptoms.114
Drug Interactions
With the exception perhaps of citalopram, and its stereoisomer escitalopram,115 SSRIs may inhibit cytochrome P (CYP) 450 isoenzymes to varying degrees, potentially causing substrate levels to rise, or reducing conversion of a substrate into its active form. Concern about drug interactions, however, is pertinent to patients who take medications with narrow therapeutic margins that are metabolized by isoenzymes inhibited by an SSRI and if the prescriber is unfamiliar with or unable to determine the appropriate dose adjustment. Given their vast availability, reports of clinically significant interactions with the SSRIs are remarkably rare. Among the SSRIs, fluvoxamine is a potent CYP 1A2 and CYP 2C19 inhibitor, and a moderate CYP 2C9, CYP 2D6, and CYP 3A4 inhibitor, while fluoxetine and paroxetine are potent CYP 2D6 inhibitors, and fluoxetine’s main metabolite, norfluoxetine, has a moderate inhibitory effect on CYP 3A4.115 Sertraline is a moderate CYP 2D6 inhibitor, while citalopram and escitalopram appear to have little effect on the major CYP isoforms.115 However, for all of the SSRIs, some vigilance is reasonable concerning the possibility of increased therapeutic or toxic effects of other co-prescribed drugs metabolized by P450 2D6. In particular, if combining a TCA with an SSRI, the TCA should be initiated with low doses, and plasma levels should be monitored. Given the high capacity of the CYP 450 3A3/3A4 system, inhibition of this isoenzyme is not a major concern for the SSRIs, although fluvoxamine, and less so fluoxetine, can inhibit it to some extent. Of little importance to drug interactions is the high rate of protein-binding of the SSRIs because, if other drugs are displaced from carrier proteins, the result is simply an increase in the rate and amount of free drug being metabolized. The augmentation and combination of SSRIs with other serotonergic agents, tryptophan, 5-HT, or MAOIs may also result in the serotonin syndrome; SSRIs should never be used concomitantly with MAOIs, because there have been a number of reports of fatal cases of serotonin syndrome due to the simultaneous use of these classes of drugs or to the inadequate washout period between the two.50–52
Use of SSRIs in Pregnancy and the Postpartum Period
There is accumulating information about the use of SSRIs in pregnancy, although the bulk of the available data is on fluoxetine. One prospective study of 128 pregnant women who took fluoxetine,116 10 to 80 mg/day (mean 25.8 mg), during their first trimester did not find elevated rates of major malformations compared with matched groups of women taking TCAs or drugs thought not to be teratogenic. There was a higher, albeit not statistically significant, rate of miscarriages in the fluoxetine (13.5%) and TCA (12.2%) groups compared with the women exposed to known nonteratogenic drugs (6.8%). Whether this increased rate of miscarriages is biologically significant and, if so, whether it relates to the drugs or to the depressive disorder could not be determined from this study. Decisions on continuing antidepressant drugs during pregnancy must be individualized, but it must be recalled that the effects of severe untreated depression on maternal and fetal health may be far worse than the unknown risks of fluoxetine or tricyclic drugs. A large registry of fluoxetine exposure during pregnancy is consistent with generally reassuring data from the TCA era that antidepressant agents are not evidently teratogens.
Serotonin Norepinephrine Reuptake Inhibitors
Venlafaxine
Venlafaxine (Effexor) was the first SNRI to gain FDA-approval for the treatment of depression. At daily doses greater than 150 mg,117 venlafaxine inhibits the reuptake of both seroto-nin and norepinephrine, while mostly inhibiting the reuptake of serotonin at lower doses.118,119 Venlafaxine lacks signi-ficant cholinergic, antihistaminergic, and alpha1-adrenergic–blocking effects. Venlafaxine is metabolized by CYP 450 2D6, of which it is also a very weak inhibitor. The half-lives of venlafaxine and its active metabolite O-desmethylvenlafaxine are about 5 and 11 hours, respectively. The drug and this metabolite reach steady state in plasma within 3 days in healthy adults. Venlafaxine is generally effective at daily doses at or above 150 mg, and is often started at 75 mg or even 37.5 mg, typically in its extended-release (XR) formulation.120 Several meta-analyses suggest venlafaxine to be more effective than the SSRIs in the treatment of MDD,121–123 with the exception of escitalopram, whose relative efficacy appears to be comparable.79
Venlafaxine, along with the SSRIs and bupropion, is also commonly chosen as a first-line treatment for depression.80 Venlafaxine is also used in treatment-refractory depression as a “next-step” strategy,124–131 reported as the most popular switch strategy for refractory depression in one large survey of clinicians.132
Common side effects of venlafaxine include nausea, insomnia, sedation, sexual dysfunction, headache, tremor, palpitations, and dizziness,133 as well as excessive sweating, tachycardia, and palpitations. There are also reports of bruxism.134,135 Venlafaxine’s potential for sexual dysfunction appears to be comparable to that of the SSRIs.136,137 The incidence of GI side effects and dizziness appears to be lower with the use of the XR formulation than the immediate-release formulation.138 Between 2% and 6% of patients also experience an increase in diastolic blood pressure,139 which appears to be dose-related.140 The abrupt discontinuation of venlafaxine, given its short half-life, also carries a risk of discontinuation-related adverse events similar to those described for the SSRIs.141 Finally, in one uncontrolled study, 4 of 13 patients treated with venlafaxine during electroconvulsive therapy (ECT) experienced asystole.142 Although the authors noted that this serious adverse event only occurred in patients on daily doses of venlafaxine greater than 300 mg and in patients anesthetized with propofol, in the absence of further data the use of venlafaxine in patients requiring ECT and perhaps even general anesthesia should be avoided.
Duloxetine
Duloxetine (Cymbalta) also inhibits the reuptake of both serotonin and norepinephrine.143 Duloxetine appears to be as effective as the SSRIs in the treatment of MDD, although in more severe depression there may be some advantages.144
Duloxetine lacks significant cholinergic, antihistaminergic, and alpha1-adrenergic blocking effects. Duloxetine is extensively metabolized to numerous metabolites that are primarily excreted into the urine in the conjugated form. The major metabolites in plasma are glucuronide conjugates of 4-hydroxy duloxetine (M6), 6-hydroxy-5-methoxy duloxetine (M10), 4,6-dihydroxy duloxetine (M9), and a sulfate conjugate of 5-hydroxy-6-methoxy duloxetine (M7). Duloxetine is metabolized by CYP 450 2D6, of which it is also a moderate inhibitor, intermediate between paroxetine and sertraline. Drugs that inhibit this enzyme may increase duloxetine concentrations. The half-life of duloxetine is about 12.5 hours. Abrupt discontinuation of duloxetine is associated with a discontinuation-emergent adverse event profile similar to that seen with SSRIs and SNRI antidepressants.145 Therefore, duloxetine’s discontinuation should be accomplished with a gradual taper (over at least 2 weeks) to reduce the risk of discontinuation-emergent adverse events. Duloxetine is commonly used at daily doses of 60 to 120 mg, often started at 30 mg. Duloxetine also appears to be effective in the treatment of somatic symptoms of depression, such as pain.146,147 Common side effects associated with duloxetine include dry mouth, headache, nausea, somnolence, sweating, insomnia, and fatigue.148 Duloxetine does not appear to cause hypertension.148
Milnacipran
A number of studies also demonstrate that the norepinephrine and serotonin reuptake inhibitor milnacipran149 is equivalent to the SSRIs150–153 and the TCAs154–158 and superior to placebo in the treatment of depression,159,160





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


