CHAPTER 63 Genetics and Psychiatry
OVERVIEW
Basic Organization of the Human Genome
The human genome comprises the full sequence of deoxyribonucleic acid (DNA) found in the nucleus of each nucleated human cell (mature erythrocytes and platelets lack nuclei and thus do not contain a copy of the genome). The DNA sequence is distributed over 23 pairs of chromosomes, long strands of DNA that include the 22 autosomes and 2 sex chromosomes. One of each pair of the autosomes and the sex chromosomes is inherited from each parent. The autosomes are numbered 1 through 22 in order of size, and most consist of two arms divided by a region called the centromere (Figure 63-1). The longer arm of a chromosome is denoted by the letter “q” and the short arm by the letter “p.” Thus, the long arm of chromosome 1 is referred to as 1q. Subdivisions of chromosomes, originally identified on the basis of chromosome staining, are referred to by numbers (e.g., 1q31.2). With the sequencing of the human genome, however, references to locations on chromosomes can now be made more precisely based on their base pair positions (e.g., a single nucleotide polymorphism at base pair 27644225 on chromosome 11).
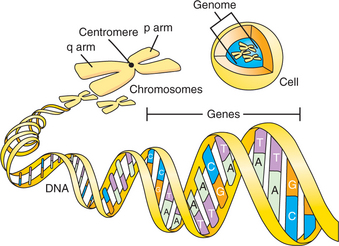
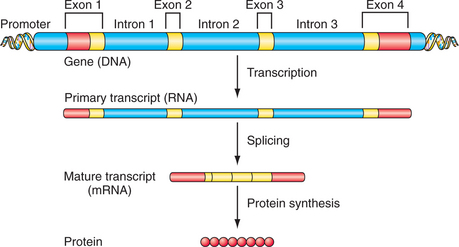
DNA encodes the instructions for making all of the proteins in the human body. Double-stranded DNA itself is composed of a linear sequence of the nucleotides adenine (A), cytosine (C), guanine (G), and thymine (T) (see Figure 63-1). The full genome sequence, comprising approximately 3 billion bases, was deciphered in 2001.1,2 Genes represent functional units of DNA sequence; the human genome contains approximately 21,000 genes. Genes convey the instruction for protein sequence through messenger ribonucleic acid (mRNA), which is transcribed from the gene sequence and, ultimately, translated into the amino acid sequence of a given protein (Figure 63-2). Protein-coding genes include protein-coding sequences (exons), intervening sequences (introns), and untranscribed regions (e.g., regulatory promoter sequences). Remarkably, however, protein-coding exons make up less than 2% of the genome sequence. Recently, evidence from a variety of studies, including the large ENCODE project, has revealed that a much larger fraction of the genome is transcribed into RNA.3
Genetic Variation and Polymorphism
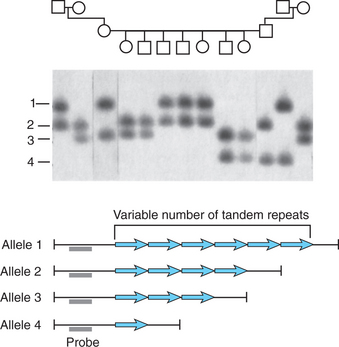
Microsatellites and variable number tandem repeats (VNTRs) are common forms of genetic variation involving short repeated sequences (Figure 63-3). Microsatellites typically comprise repeats of two to four nucleotides (e.g., CA repeats). These repeat sequences are typically found outside of the amino acid coding sequences, but their high degree of polymorphism has made them useful as genetic markers in genetic linkage studies. In some cases, short repeats do have functional effects. One of the best known examples relevant to psychiatry is the repeat sequence in the promoter of the serotonin transporter gene (the so-called 5HTT promoter length polymorphism or 5HTTLPR). The “short” allele of the 5HTTLPR, lacking 44 base pairs of repeat sequence that are present in the “long” allele, confers reduced transcriptional activity of the serotonin transporter gene and has been associated with a variety of neuropsychiatric phenotypes (see below).
Variable numbers of repeated three base-pair sequences, known as triplet repeats, have been shown to play a role in several neurological illnesses. For example, CAG repeats (which encode the amino acid glutamine) within the Huntingtin gene on chromosome 4 cause Huntington disease when more than 40 repeats are present (Figure 63-4). Alleles containing fewer than 35 repeats do not produce disease, but as repeat length increases, instability in replication can lead to expansion of the repeat sequence. This accounts for the phenomenon of anticipation in which the mutation “worsens” over successive generations, resulting in earlier-onset and more severe disease phenotypes. Other diseases associated with triplet repeat expansion include fragile X, spinocerebellar ataxias, and myotonic dystrophy.

Single nucleotide polymorphisms (SNPs), in which one of the four nucleotide bases is substituted for another, are the most common form of genetic variation, occurring approximately once per 1,000 base pairs (bp) of DNA sequence. Because they are so common (approximately 10 million in the genome) and may have functional significance, SNPs have become the focus of genetic association analysis, the most widespread approach to identifying susceptibility genes for complex disorders (see below). SNPs may affect phenotypic variation through several mechanisms. One straightforward mechanism results when an SNP in the exon of a gene alters or disrupts the instructions for the normal amino acid sequence of the gene product. Such “nonsynonymous” coding sequence SNPs can result in an abnormal or truncated protein product with aberrant or no function. There is now evidence that “synonymous” or “silent” SNPs that alter the coding sequence but do not result in an amino acid change can also have functional effects (e.g., by leading to more subtle changes in protein conformation).4 In addition, SNPs occurring in the regulatory regions of genes (e.g., the promoter) can induce phenotypic effects through alterations in gene expression.
Recently, the widespread occurrence of copy number variation in the genome has been reported.5 These variations include deletions, insertions, and duplications of DNA sequence, ranging from kilobases (thousands of bases) to megabases (millions) in length, that may alter gene function or dose. The extent and frequency of copy number variants and their relationship to complex disease is an area of active research. Examples of copy number variation relevant to psychiatry include the 22q11 microdeletion that causes velocardiofacial/DiGeorge syndrome and is associated with psychotic illness,6 the duplication or deletion of CYP450 2D6 genes that can alter psychotropic drug metabolism,7 and the recent report of copy number variants associated with autism.8
Linkage Disequilibrium and Haplotypes
The phenomenon of linkage disequilibrium (LD) is an important characteristic of the human genome. LD refers to the correlation or association of alleles at linked polymorphic markers. A key feature of the structure of the human genome is that it is organized into regions of high LD separated by regions of low LD. Markers that exhibit high levels of LD and that reside on the same chromosome (i.e., markers whose alleles are strongly correlated) are referred to as haplotypes. Put another way, a haplotype refers to a set of strongly associated alleles at markers along a chromosomal region that tend to be inherited together.9 LD arises when a new variant occurs on a chromosome (e.g., through mutation). The chromosome on which the variant arises is surrounded by other alleles and so it is inherited along with those alleles in the subsequent generation. Over successive generations, recombination of chromosomes and other mutations at neighboring sites dilute the degree of correlation between the new variant allele and surrounding alleles, degrading the extent of LD between them. However, human populations are relatively young so that stretches of LD have persisted. In regions of high LD, there is limited haplotype diversity—that is, only a few haplotypes exist in the population in these regions. A major advance in cataloguing genetic variation and LD across the human genome was accomplished by the International HapMap Project.10 The HapMap Project examined the frequencies and LD relationships of more than a million SNPs across the genome in multiple populations: Yoruban Africans, Caucasian Americans, Han Chinese, and Japanese. Detailed analyses confirmed that the genome comprises long stretches of LD with limited haplotype diversity interrupted by regions of low LD (in “recombination hotspots”). As expected, these analyses also revealed important population differences in allele frequencies and in the length of LD blocks (with the shortest blocks observed in the Yoruban population due to the ancestral origin of the human species in Africa and thus a greater opportunity for decay of LD over successive generations). A crucial dividend of the HapMap is its utility for genomewide association studies (described below).
Gene Expression
Gene expression refers to the process and products of gene transcription into RNA and, if applicable, translation into protein (see Figure 63-2). The regulation of gene expression involves both genetic and epigenetic factors. Genetic influences on expression include DNA sequences in regions around genes known as promoters and enhancers. These regulatory regions function in part by serving as binding sites for transcription factors that help determine where (in what tissues) or when (during which developmental periods) genes are activated or silenced. Sequence variations in regulatory regions provide another mechanism (beyond variations in protein coding regions of genes) by which DNA sequence differences contribute to phenotypic differences among individuals and populations.11
Another important mechanism for modulation of gene expression involves epigenetic regulation. Epigenetics refers to the study of heritable gene expression changes that are not due to variation in DNA sequence.12 Chromatin, the complex of DNA, histones, and associated nonhistone proteins, is the primary substrate of epigenetic modulation. Histones are highly basic proteins found in the nucleus of the cell; chromatin consists primarily of DNA molecules wrapped around octomeric complexes of histones.13 The configuration of chromatin can vary from states in which it is inactivated and condensed (also known as heterochromatin) or activated and open (also known as euchromatin).13 Genes in regions of condensed chromatin are inaccessible to transcription factors and thus functionally repressed, while those in regions of open chromatin are available to transcriptional activation. Shifts in chromatin structure (chromatin remodeling) may thus lead to important variations in gene expression with downstream effects on a variety of phenotypes. The chemical and molecular bases of chromatin remodeling and epigenetic modification of gene expression include histone acetylation (which generally increases transcriptional activity), histone methylation (which can increase or decrease transcriptional activity), and DNA methylation (which generally reduces transcriptional activity). A variety of factors appear to influence epigenetic modification of chromatin and DNA, including aging, stress, diet, and various drugs and medications.12–15 Indeed, modification of the epigenome may be a central mechanism by which environmental influences are transduced into molecular effects on gene expression and action.
A number of neuropsychiatric disorders and phenotypes have been linked to epigenetic variations, including Rett syndrome (an autism spectrum disorder caused by mutations in the transcriptional repressor MeCP2), depression, schizophrenia, and addiction.13
Finally, gene expression can also be regulated by RNA interference (RNAi) due to noncoding RNAs, including short interfering RNA (siRNA), microRNA (miRNA), and small hairpin RNA (shRNA). These RNAs can modulate gene expression by several mechanisms, including degrading mRNAs of target genes or interfering with translation of mRNA into proteins.16 The role of these factors in human disease is an area of active investigation.
Complex versus Mendelian Genetics
The genetic basis of medical disorders is often categorized as either “Mendelian” or “complex.” The prototypic Mendelian disorder is one in which mutations or alterations in a single gene cause a disease according to classical Mendelian patterns of inheritance (e.g., recessive, dominant, or X-linked) and are often highly penetrant (i.e., the risk of illness in those carrying the genetic liability is high). Dominant inheritance occurs when variations in a single copy of the relevant gene are sufficient to cause disease, and recessive inheritance applies when mutations in both copies of a gene are needed. In contrast, complex phenotypes, which include common medical and psychiatric disorders, reflect the additive or interactive influence of multiple genes and environmental influences. In some cases, single major genes (genes of relatively large effect) may contribute along with several other genes (oligogenic inheritance). Polygenic inheritance involves the cumulative effects of many genes of individually small effect. Most common medical and psychiatric disorders are believed to have multifactorial etiologies, involving both genetic and environmental (and possibly epigenetic) factors. In many cases, gene-gene interaction (epistasis) and gene-environment interaction may be essential to the expression of complex disease phenotypes. As more is learned about the genetic basis of single gene disorders, the distinction between complex and Mendelian disorders is increasingly difficult to draw. For example, even for a paradigmatic Mendelian disorder such as Huntington disease, additional genes may regulate the course and age of onset of disease.17
Recent studies in diverse areas of medicine have supported the hypothesis that common, complex disorders reflect the action of multiple genes of small individual effect. Using genomewide association methods (see next), several genes have been identified that confer susceptibility to common medical disorders, including diabetes,18–20 coronary artery disease,19,21 Crohn’s disease,22,23 and other complex diseases; however, the individual effects of these susceptibility variants are small (with odds ratios typically less than 1.5), and many thousands of cases and controls may be required to detect such effects. Nevertheless, some proportion of cases may be attributable to rarer, more highly penetrant variants that have not yet been detected.
Approaches to the Study of Psychiatric Genetics
Genetic Epidemiology
Family Studies.
Family studies address the first question to be answered: Does the disorder run in families? The design of a typical family study is similar to other case-control studies. Cases (affected probands) and controls (unaffected probands) are ascertained and the lifetime prevalence of the disorder is measured among their (usually first-degree) relatives. A higher prevalence among relatives of affected probands is evidence that the disorder aggregates in families. The risk to relatives of affected probands is referred to as the “recurrence risk.” One index of the strength of familiality is the “recurrence risk ratio” for first-degree relatives (λ1), defined as the ratio of the risk of the disorder in a first-degree relative of an affected individual to the prevalence in the general population. It is important to bear in mind that the size of these risk ratios depends on both the risk to relatives (numerator) and the base rate of the disorder (denominator). Even when the relative risk of a disorder is high, the absolute risk to a first-degree relative may be relatively low if the base rate of the disorder is low. For example, siblings of probands with schizophrenia have a roughly tenfold increased risk of the disorder compared to an individual randomly drawn from the general population (λ1 ≈ 10). However, because the population prevalence is approximately 1%, the absolute risk of the disorder for the sibling is only about 10% (with a 90% probability of being unaffected). In contrast, the lifetime prevalence of major depression is approximately 15%, so that even a twofold increased risk to siblings would be associated with a 30% risk of being affected. Family studies can also provide information about the etiological boundaries or relatedness of different traits or diagnoses. For example, relatives of probands with Tourette syndrome have an elevated risk of obsessive-compulsive disorder (OCD), suggesting that these conditions have overlapping familial determinants (Table 63-1).
Methodological issues can influence the interpretability of family studies. Studies using the “family history method” rely on informant reports to assign diagnoses (e.g., probands may be interviewed about their relatives). Because this method can be less sensitive than direct-interview methods for detecting psychopathology in relatives,24 the latter are considered the gold standard. The “family study method” involves direct assessment of probands and relatives, although informant reports may be incorporated to derive “best-estimate” diagnoses using all available data.
Twin Studies.
Linkage Analysis.
If individuals affected with a disorder within a family tend to inherit the same alleles at a marker locus, this implies that the marker locus is linked to (i.e., is physically close to) a gene that influences the disorder. In classical (parametric) linkage analysis, the strength of the evidence in favor of linkage is calculated as a logarithm of the odds (LOD) score. The LOD score compares the likelihood of obtaining the observed genotypes and phenotypes when linkage is present with the likelihood assuming no linkage. For classic single gene (Mendelian) disorders, a LOD score of 3 (corresponding to odds of 1,000 : 1 in favor of linkage) has been the threshold for declaring linkage; for complex disorders, such as psychiatric illnesses, higher thresholds (3.3 to 4.0) have been recommended.26 Traditional LOD score linkage analysis requires that a model including several parameters (mode of inheritance, disease allele frequencies, and marker allele frequencies) be specified. Thus, linkage analysis has been most successfully applied when a single major gene is involved and the mode of inheritance (e.g., dominant, recessive) is known. Because these parameters are rarely known with psychiatric phenotypes, other (“model-free”) methods that do not require knowledge of mode of inheritance are routinely used. For example, genome scan linkage analysis may be performed with large numbers of families containing pairs of relatives who are affected. The affected sibling pair (ASP) method, for example, examines whether affected siblings share alleles at any marker locus significantly more often than would be expected by chance. If alleles at a locus tend to be shared by affected sibling pairs, this implies that the locus is linked to a disease gene.
Association Analysis.
Whereas linkage analysis examines the co-inheritance of alleles and phenotypes within families, association analysis examines the co-inheritance of alleles and phenotypes across families (i.e., across a population of unrelated individuals) (Figure 63-5). While linkage analysis asks “where” a susceptibility gene resides, association analysis asks “which” specific genetic variants influence a phenotype. In recent years, association methods have increasingly replaced linkage methods for the study of complex diseases. This is because association methods are more powerful than linkage analysis for detecting small genetic effects. However, association between variants operates over much shorter genomic distances, so that much denser sets of genetic markers are needed.
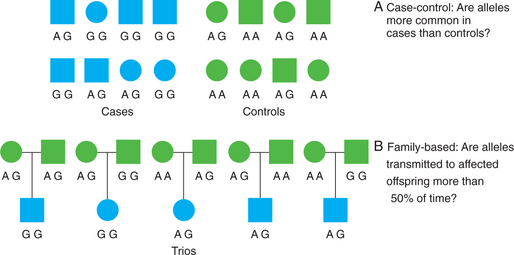
Figure 63-5 Design of genetic association studies showing case-control (A) and family-based (B) association methods.
Confounding due to population stratification or admixture can occur when cases and controls differ in their population genetic backgrounds. As noted previously, allele frequencies often differ between different populations. Thus, if case and control groups differ in the proportion of individuals from different populations or ethnicities, any differences in allele frequencies may simply be due to population differences rather than true genetic effects on disease status. Several methods have been developed to avoid this type of confounding, including genomic control and structured association methods.27 Another way to avoid confounding by population stratification is to use family-based association tests that look at allele transmission within families.28 The paradigmatic family-based association test is the transmission/disequilibrium test (TDT). This test examines transmissions of alleles from heterozygous parents to affected offspring. Under the null hypothesis of no association and no linkage, there is a 50% probability that a heterozygous parent will transmit one of two alleles to her offspring. If a given allele is transmitted to offspring affected with the trait of interest significantly more than 50% of the time, this is evidence that the allele is directly or indirectly associated with the trait.
Candidate Gene Studies
While some candidate genes have shown evidence of association in multiple studies, findings have generally been inconsistent and nonreplications are common. This likely reflects several limitations of candidate gene association studies. First, while all brain-expressed genes are plausible candidates, the evidence to date for most genes is not compelling. Because the neurobiological basis of psychiatric disorders remains poorly understood, the prior probability that any particular candidate gene will be associated with a given disorder is low. This increases the likelihood that association findings for such candidates are spurious false-positive results due to chance.29 Candidate gene studies to date have also commonly been underpowered. It is increasingly clear that very large sample sizes (on the order of thousands of cases and controls) are needed to detect the small effects that genes underlying complex traits are likely to exert. Thus, negative findings can be uninformative if power is inadequate. Similarly, studies that examine a limited number of genetic variants within a gene may fail to detect true association that would have been evident with a more comprehensive analysis of polymorphisms.
Genomewide Association Studies
In recent years, advances in high-throughput genotyping technologies coupled with the extensive cataloguing of genetic variation through the International HapMap Project have made genomewide association analysis possible. These studies make use of the fact that there is extensive LD across the genome so that the alleles of many SNPs are strongly correlated. Thus, a given SNP may provide information about other (correlated) SNPs, so that common genetic variation throughout the genome can be assayed without having to genotype all of the millions of individual SNPs that actually exist. By selecting a reduced set of SNPs that efficiently “tag” nearby genetic variants, the entire genome can be interrogated using DNA chips that simultaneously assay 500,000 to 1,000,000 SNPs.30 Such genotyping technology is now available and has been used to identify genes influencing common, complex disorders. Unlike candidate gene studies, genomewide association studies are “unbiased” in that they do not require a prespecified hypothesis about which genes are important; as such, they offer the opportunity to uncover novel “players” in the biology of disease risk or response to treatment.
Gene-Environment Interaction
Genes operate in the inextricable context of environments, and the etiology of psychiatric illness is generally believed to involve additive and interactive effects of genetic susceptibility and environmental stressors. Despite this belief, most research to date has focused either on genetic or environmental risk factors without examining how these influences act in concert. One reason for the inconsistency in genetic association findings may be that risk alleles may only have observable effects in the context of specific environmental exposures. Thus, failure to measure and incorporate environmental factors may obscure genotype-phenotype relationships.31 More recently, the examination of gene-environment interaction has become a major focus of research, in part because genetic association methods are well-suited to examine additive and interactive effects of multiple risk factors. Ironically, it has become a simpler matter to measure genetic variation than environmental risk factors: the genome is finite but the environment is nearly unbounded. Identifying which candidate environmental factors are relevant and capturing their longitudinal effects can be difficult. In addition, the sample sizes required for adequate power to examine interactions may be substantially larger than those needed to study main effects. Despite these challenges, a growing number of studies are documenting allele-environment interactions for com-mon psychiatric phenotypes. Caspi and colleagues32 initially reported two such interactions that have been replicated in several (though not all) independent samples: an interaction between the “short” allele of the serotonin transporter 5HTTLPR polymorphism and stressful life events conferring risk for depression and an interaction between a functional variant in the MAO-A gene and childhood maltreatment conferring risk for antisocial behavior.33 Several reviews of the scientific and methodological issues in conducting such studies in psychiatric research are available.34,35
Intermediate Phenotypes and Endophenotypes
Gottesman and Gould36 highlighted five desirable characteristics of a putative endophenotype:
A large number of endophenotypes and intermediate phenotypes have been proposed for psychiatric disorders, though data regarding all five criteria listed above are not available for many of these. An example of an endophenotype that appears to meet most of the criteria is inhibition of the P50 evoked response to repeated auditory stimuli, a phenotype that may underlie the abnormalities in sensory gating observed in schizophrenia.37 A deficit in P50 inhibition has been associated with schizophrenia and the phenotype appears to be heritable and to co-segregate with the illness in families.38,39 Linkage of impaired P50 inhibition was reported to a locus on chromosome 15q, adjacent to the alpha-7 nicotinic receptor gene,40 and subsequent analyses provided evidence that variants in the promoter of this gene are associated with both P50 inhibition and schizophrenia.41
Neuroimaging phenotypes are attractive for genetic studies because they directly measure brain structure or function. The growing literature on imaging genetics42 has identified several genotype-phenotype correlations involving specific genetic variants. For example, the functional promoter polymorphism in the serotonin transporter gene (5HTTLPR) has been associated with increased amygdala reactivity and reduced coupling of corticolimbic circuits, and neuroimaging phenotypes have been implicated in the biology of anxiety and depressive disorders.43,44 Other studies have implicated functional polymorphisms in catechol O-methyltransferase (COMT) and prefrontal cortical phenotypes have been thought to underlie working memory deficits and dopaminergic dysregulation in schizophrenia.42,45 Nevertheless, a recent meta-analysis of such studies challenged the claim that endophenotypes will reveal larger genetic effects than those observed with traditional psychiatric diagnoses.46
GENETICS OF PSYCHIATRIC DISORDERS
Disorders of Childhood and Adolescence
Attention-Deficit/Hyperactivity Disorder
Genetic Epidemiology.
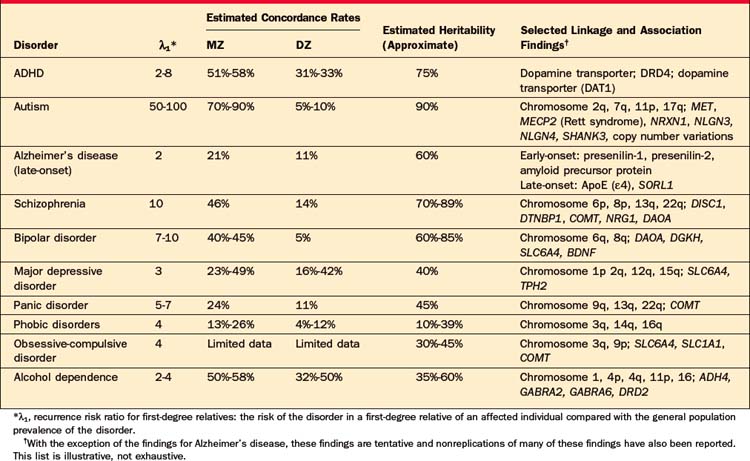
Numerous family studies have demonstrated that attention-deficit/hyperactivity disorder (ADHD) runs in families. First-degree relatives (parents and siblings) of ADHD probands have a twofold to eightfold higher risk of the disorder than relatives of controls.47 Family studies also suggest that ADHD and depression share familial determinants,48 and that ADHD with conduct or bipolar disorder may be a distinct familial subtype.49–51 Most twin studies have shown significantly higher concordance rates for MZ as compared with DZ twins. The mean heritability estimate from 20 twin studies is 76%.47 Adoption study data for ADHD are quite limited, and early studies had methodological shortcomings; a small adoption study showed higher rates of ADHD in biological compared with adoptive relatives of ADHD probands.52
Molecular Genetic Studies.
Genomewide linkage studies of ADHD have provided evidence implicating a number of chromosomal regions. A few regions have shown evidence of linkage in multiple genome scans including 16p13 and 17p11.53–57 A large number of candidate gene studies have been reported for ADHD. Many of these studies have focused on genes involved in dopaminergic neurotransmission based on the observation that stimulants, the most widely used pharmacotherapy for ADHD, act to block dopamine reuptake. Meta-analyses of candidate gene studies have reported significant, though modest, evidence of association for four dopaminergic genes [the dopamine transporter (SLC6A3), the dopamine 4 receptor (DRD4), dopamine beta hydroxylase (DBH), and the dopamine 5 receptor (DRD5)]; two serotonergic genes [the serotonin 1B receptor (HTR1B) and the serotonin transporter (SLC6A4)]; and a gene involved in exocytotic neurotransmitter release (SNAP-25).47,58,59 Genomewide association studies of ADHD are currently underway.
Autism
Genetic Epidemiology.
The risk of autism to siblings of affected children is approximately 2%-7%, which is 50 to 100 times higher than the general population prevalence.60,61 When the “broader autism phenotype” (including autism spectrum disorders and milder abnormalities of social and language function) is considered, the risk to first-degree relatives may be as high as 10% to 45%.61 Concordance rates for MZ twins are markedly higher (70% to 90%) than those for DZ twins (5% to 10%), and the heritability has been estimated to exceed 90%.62
Molecular Genetic Studies.
More than 10 whole genome linkage studies have been reported for autistic disorder in the past decade. A region on chromosome 7q has shown evidence of linkage in most scans and linkage to the 7q22-q32 region was supported in a meta-analysis of genome scans.63 Regions on chromosome 2q and 17q have also received support in more than one linkage analysis. As in other complex disorders, it is likely that multiple genes of varying effect contribute to common forms of autism. A large scale SNP-based linkage study has implicated the 11p12-p13 region.64 A number of candidate genes have been associated with autism or autism spectrum phenotypes, including the serotonin transporter gene (SLC6A4), engrailed 2 (EN2), and a functional variant in the promoter of the MET protooncogene.65,66 A subset of autism cases may be attributable to copy number variations (small duplications or deletions of genomic sequence or genes) resulting from de novo mutations.8 Cytogenetic studies have also implicated duplications on chromosome 15q in some cases. Autism or autistic symptoms also occur in several medical genetic disorders for which specific genes have been identified, including neurofibromatosis (the NF1, NF2I genes), tuberous sclerosis (TSC1, TSC2), fragile X (FMR1), Rett syndrome (MECP2), and others.60 Recent studies have also implicated genes involved in synapse formation and function. For example, mutations or disruptions in genes encoding neuroligins and neurexins and their binding partner SHANK3, key components in synapse mutation and function,67 have been detected in cases of autism.64,68,69 Genomewide association studies are underway. The finding that advanced parental age is associated with risk of autism70 supports the view that a subset of cases are due to de novo germline mutations (which are more likely to occur with advancing age). Zhao and colleagues71 have proposed that most cases of autism result from de novo mutations in the parental germ lines that are highly penetrant in male offspring but less penetrant in females (for as yet unknown reasons). However, male children of these female offspring would inherit the mutations with 50% probability and develop autism with high probability.
Tourette Syndrome
Genetic Epidemiology.
Familial aggregation studies have found an approximately fivefold to fifteenfold increased risk in first-degree relatives of Tourette syndrome (TS) probands compared to the general population (7% to 18% vs. 1% to 2%, respectively).72 There is evidence for variable expression of the genetic liability for TS; for example, relatives of probands with TS have a higher risk of OCD, and chronic motor or vocal tics.73 Concordance rates in MZ twins (50% to 70%) are significantly greater than rates in DZ twins (9%).72
Molecular Genetic Studies.
Genetic linkage studies have provided supportive evidence for several chromosomal regions. The largest of these included 304 affected sib-pair families and 18 large multigenerational families and found significant evidence of linkage on chromosome 2p.74 Several specific genes have been implicated in association and cytogenetic studies, though none has been established.72 Variants in the slit and Trk-like 1 gene (SLITRK1) may be a rare cause of TS.75
Dementia: Alzheimer’s Disease
Genetic Epidemiology.
The familiality of early-onset (before age 60 to 65 years) Alzheimer’s disease (AD) has been well established, and three specific genes influencing early-onset AD have been identified (see below). Inheritance of early-onset AD follows an autosomal dominant pattern, but the early-onset form is rare, with a prevalence under 0.1%.76 Late-onset AD is far more common and has a more complex etiology. Having an affected first-degree relative is associated with an approximately 2.5-fold increased risk of AD.77 Twin studies have estimated the heritability of late-onset AD at 48% to 60%.78–80
Molecular Genetic Studies.
Mutations in three genes have been shown to produce early-onset AD with an autosomal dominant mode of inheritance: the amyloid precursor protein (APP) gene on chromosome 21, presenilin 1 (PS1) on chromosome 14, and presenilin 2 (PS2) on chromosome 1. Together, these genes account for roughly half of early-onset cases of AD, but less than 5% of AD cases overall.81 The apolipoprotein E gene (APOE) on chromosome 19 is an established risk factor for late-onset AD. There are three common alleles of APOE (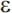 2,
2, 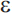 3, and
3, and 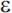 4), and it is the
4), and it is the 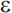 4 allele that increases risk for late-onset AD (APOE-
4 allele that increases risk for late-onset AD (APOE-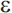 2 appears to be associated with a reduced risk of AD). Unlike the autosomal dominant genes involved in early-onset AD, APOE-
2 appears to be associated with a reduced risk of AD). Unlike the autosomal dominant genes involved in early-onset AD, APOE-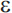 4 is a susceptibility allele that acts as a risk factor for the disease but is neither a necessary nor sufficient cause. A primary effect of the
4 is a susceptibility allele that acts as a risk factor for the disease but is neither a necessary nor sufficient cause. A primary effect of the 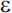 4 allele is to reduce the age of onset of AD82; individuals with two copies of the allele have the earliest age of onset compared to individuals with other APOE genotypes. Several other genes have compelling evidence for involvement in AD, including SORL1.83 A systematic meta-analysis of association studies identified 13 genes with evidence of significant association84; an online database of association studies and meta-analytic results (AlzGene) is maintained at www.alzforum.org. Of note, as in other complex disorders, susceptibility genes for late-onset AD appear to have modest effects on risk (odds ratio [OR] in the range of 1.1 to 1.4). Finally, the high risk of AD among individuals with Down syndrome (trisomy 21) has been attributed largely to triplication of the APP gene on chromosome 21. Onset of AD in these individuals is typically in the sixth decade of life.
4 allele is to reduce the age of onset of AD82; individuals with two copies of the allele have the earliest age of onset compared to individuals with other APOE genotypes. Several other genes have compelling evidence for involvement in AD, including SORL1.83 A systematic meta-analysis of association studies identified 13 genes with evidence of significant association84; an online database of association studies and meta-analytic results (AlzGene) is maintained at www.alzforum.org. Of note, as in other complex disorders, susceptibility genes for late-onset AD appear to have modest effects on risk (odds ratio [OR] in the range of 1.1 to 1.4). Finally, the high risk of AD among individuals with Down syndrome (trisomy 21) has been attributed largely to triplication of the APP gene on chromosome 21. Onset of AD in these individuals is typically in the sixth decade of life.
Psychotic Disorders: Schizophrenia
Genetic Epidemiology.
Family studies of schizophrenia have repeatedly demonstrated that the disorder is familial. Compared to the population lifetime risk of approximately 1%, first-degree relatives have an approximately 10% risk. The risk drops to about 4% for second-degree relatives and 2% for third-degree relatives.85 There is evidence for variable expression of the genetic diathesis underlying schizophrenia. For example, schizoaffective disorder and cluster A personality disorders are more common in the relatives of schizophrenic probands.86 The concordance rate for MZ twins (approximately 50%) substantially exceeds that of DZ twins (approximately 15%).86 The heritability of schizophrenia has been estimated to be 70% to 89%.86,87 Adoption studies have demonstrated that the prevalence of schizophrenia is significantly higher (approximately fourfold) in biological relatives than in adoptive relatives.86
Molecular Genetic Studies.
Twenty-eight whole genome scan linkage studies of schizophrenia have been reported and have implicated numerous chromosomal regions.88 In a meta-analysis of 20 genome scans, Lewis and associates89 reported genomewide significant evidence of linkage on chromosome 2q and strong evidence for regions on chromosomes 1q, 3p, 5q, 6p, 8p, 11q, 14p, 20q, and 22q. Psychotic disorder phenotypes associated with cytogenetic abnormalities have provided additional strong evidence for regions of chromosomes 1q90,91 and 22q.6 Chromosomal microdeletions in the 22q11 region have been associated with velocardiofacial syndrome (see below), in which affected individuals appear to have an excess risk of schizophrenia and schizoaffective disorder. Some of these regions contain candidate loci that have been associated with schizophrenia in several studies. These include RGS4 and DISC1 on chromosome 1q, dysbindin (DTNBP1) on 6p, neuregulin 1 (NRG1) on 8p, and PRODH and catechol O-methyltransferase (COMT) on 22q.92 A database of association study results and meta-analyses can be found at www.schizophreniaforum.org/res/sczgene. Other genes implicated in schizophrenia susceptibility include d-amino acid oxidase activator (DAOA),93 and the gene encoding 5,10 methylenetetrahydrofolate reductase (MTHFR).94 A whole genome association study implicated cytokine-related genes (CSF2RA and IL3RA) in the pseudoautosomal region of the X and Y chromosomes.95 Ongoing whole genome association analyses are likely to provide additional putative susceptibility genes.
Mood Disorders
Bipolar Disorder
Genetic Epidemiology.
Data from nearly 20 family studies have documented that bipolar disorder (BPD) is familial.96 Overall, a summary estimate of familial risk indicates that the recurrence risk of BPD for first-degree relatives of bipolar probands is approximately 10% (recurrence risk ratio approximately 10), while the risk for unipolar major depressive disorder (MDD) is approximately 15% to 20% (recurrence risk ratio approximately 2 to 3). Increased familial risks have been associated with early-onset BPD.96 Concordance rates are substantially higher in MZ twins (approximately 40% to 45%) than in DZ twins (approximately 5%), and the heritability of BPD has been estimated to be approximately 60% to 85%.97–100 The two adoption studies that have used a modern definition of BPD101,102 were too small to support clear conclusions about the heritability of BPD itself. Twin studies also suggest that genetic influences on BPD overlap with those contributing to MDD, schizophrenia, and schizoaffective disorder.99,103
Molecular Genetic Studies.
More than 40 linkage studies of BPD have been reported. Suggestive evidence of linkage has been reported for almost all chromosomes, but few genomewide significant results have emerged. Three meta-analyses, combining results of BPD genome scans, have yielded conflicting results.104–106 The most comprehensive meta-analysis identified genomewide significant evidence for linkage on chromosomes 6q (for bipolar I disorder) and 8q (for bipolar I + bipolar II disorder).105 A large number of candidate gene studies have been reported, though with few replicated findings.107 Multiple studies or meta-analyses of association studies have supported a role for SLC6A4,108 BDNF,109–111 DAOA,93 and MTHFR.94 Several of the genes with the strongest evidence of association for BPD have also been strongly implicated in susceptibility to schizophrenia, supporting the evidence from twin studies that genetic influences on these disorders overlap in part.112 Whole genome association studies of BPD have begun to appear. Initial results have implicated diacyl glycerol eta (DGKH) on chromosome 13q113 and a variant near several plausible candidate genes on 16p.19
Major Depressive Disorder
Family Studies.
A substantial body of evidence has established that MDD is a familial phenotype, with family studies estimating recurrence ratios ranging from approximately twofold to ninefold.114 A meta-analysis of family studies found that the prevalence of MDD was threefold higher in the relatives of affected probands compared to the relatives of unaffected controls (summary OR = 2.84; 95% CI: 2.31 to 3.49).115 Certain features of MDD in the proband have been associated with increased familial risk: early onset, recurrent episodes, chronicity, suicidality, and greater levels of impairment.116–122 In twin studies published since 1985, the MZ concordance rates have typically fallen in the range of 30% to 50% while DZ concordance rates have typically ranged from 12% to 40%, with somewhat higher rates seen in female compared with male twin pairs.115,123 Combining these studies, Sullivan and colleagues115 estimated the summary heritability at 37% (95% CI: 33% to 42%), with a larger share of the variance explained by individual-specific environment (63%, 95% CI: 58% to 67%). The absence of a significant effect of shared family environment suggests that the familial aggregation of MDD is due mostly or entirely to genetic influences. These estimates are consistent with those of the largest twin study comprising more than 15,000 Swedish twin pairs in which the heritability of MDD was estimated at 42% for women and 29% for men.124 The results of four available adoption studies have been mixed, providing modest support for a genetic component.114
Molecular Genetic Studies.
Genetic linkage studies of MDD have implicated several chromosome regions as harboring susceptibility genes, including 1p,125–128 2q,129 12q,125,130 and 15q,125,131 though these await confirmation. A recent large linkage study of families with recurrent early-onset MDD provided strong support for the 15q region,132,133 and fine-mapping of the region indicated that a 15q25-q26 locus increases risk to siblings by up to 20%.133 Of the many candidate genes examined, the most widely studied has been the gene encoding the serotonin transporter (SLC6A4) on chromosome 17q, the therapeutic target of selective serotonin reuptake inhibitor (SSRI) antidepressants. In particular, a common polymorphism in the promoter of the serotonin transporter gene (the serotonin transporter length polymorphic region, 5HTTLPR) has been the focus of association studies in mood disorders and other mood disorder phenotypes.134 As noted earlier, two common alleles exist and are distinguishable by the insertion (“long” allele) or deletion (“short” allele) of a 44 base pair sequence.135,136 The “short” allele has been associated with reduced expression of the serotonin transporter. Several meta-analyses of association studies of this polymorphism in MDD have been reported, with mixed results.137–139 More consistent results have been obtained in studies that have incorporated analyses of gene-environment interaction. Caspi and colleagues32
Related posts:
 31: Psychiatric Illness during Pregnancy and the Postpartum Period
32: Anxiety Disorders: Panic, Social Anxiety, and Generalized Anxiety
7: Understanding and Applying Psychological Assessment
59: Approaches to Collaborative Care and Primary Care Psychiatry
85: The Role of Psychiatrists in the Criminal Justice System
76: Seizure Disorders (Epilepsy)
31: Psychiatric Illness during Pregnancy and the Postpartum Period
32: Anxiety Disorders: Panic, Social Anxiety, and Generalized Anxiety
7: Understanding and Applying Psychological Assessment
59: Approaches to Collaborative Care and Primary Care Psychiatry
85: The Role of Psychiatrists in the Criminal Justice System
76: Seizure Disorders (Epilepsy)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


