CHAPTER 50 Drug-Drug Interactions in Psychopharmacology
OVERVIEW
An understanding of drug-drug interactions is essential to the practice of psychopharmacology.1,2 As in other areas of medicine, polypharmacy has become an increasingly accepted approach in psychiatry for addressing difficult-to-treat disorders.3 Moreover, general medical co-morbidity is common among patients with psychiatric disorders, elevating the likelihood of complex medication regimens.4,5 Similarly, the widespread use of over-the-counter (OTC) supplements by patients receiving treatment for psychiatric disorders may invite additional risk of drug-drug interactions.6 When they occur, drug-drug interactions may manifest in myriad ways, from perplexing laboratory tests to symptoms that are difficult to distinguish from the underlying psychiatric and physical conditions under treatment. The comprehensive evaluation of patients with psychiatric disorders therefore requires a careful assessment of potential drug-drug interactions.
Drug-drug interactions refer to alterations in drug levels or drug effects (or both) attributed to the administration of two or more prescribed, illicit, or OTC agents in close temporal proximity. Although many drug-drug interactions involve drugs administered within minutes to hours of each other, some drugs may participate in interactions days or even weeks after their discontinuation because of prolonged elimination half-lives (e.g., fluoxetine) or due to their long-term impact on metabolic enzymes (e.g., carbamazepine). Some drug-drug interactions involving psychotropic medications are life-threatening, such as those involving the co-administration of monoamine oxidase inhibitors (MAOIs) and drugs with potent serotonergic (e.g., meperidine) or sympathomimetic (e.g., phenylpropanolamine) effects.7–9 These combinations are therefore absolutely contraindicated. However, most drug-drug interactions in psychopharmacology manifest in somewhat more subtle ways, often leading to poor medication tolerability and compliance due to adverse events (e.g., orthostatic hypotension, sedation, or irritability), diminished medication efficacy, or puzzling manifestations (such as altered mental status or unexpectedly high or low drug levels). Drug combinations that can produce these often less catastrophic drug-drug interactions are usually not absolutely contraindicated. Some of these combinations may, indeed, be valuable in the treatment of some patients though wreaking havoc for other patients. The capacity to anticipate and to recognize both the major, but rare, and the more subtle, but common, potential drug-drug interactions allows the practitioner to minimize the impact of these interactions as an obstacle to patient safety and to therapeutic success. This is both an important goal and a considerable challenge in psychopharmacology.
While drug-drug interactions are ubiquitous, few studies have systematically assessed in vivo drug-drug interactions of most interest to psychiatrists. Fortunately, well-designed studies of drug-drug interactions are an increasingly integral part of drug development. Beyond these studies, however, the literature on drug-drug interactions remains a patchwork of case reports, postmarketing analyses, extrapolation from animal and in vitro studies, and extrapolation from what is known about other drugs with similar properties. While these studies often shed some light on the simplest case of a single drug (drug B) exerting an effect on another (drug A), they rarely consider the common clinical scenario in which multiple drugs with numerous potential interactions among them are co-administered. Under these circumstances, the range of possible, if not well-delineated, drug-drug interactions often seems overwhelming.
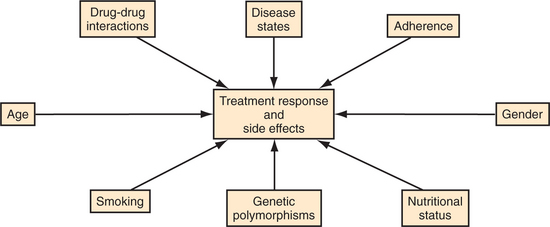
Fortunately, an increasing range of resources are available (including prescribing software packages and regularly updated Web sites such as www.drug-interactions.com) that allow for the prevention and detection of potential interactions. In addition, it is important to recall that numerous factors contribute to inter-individual variability in drug response.10,11 These factors include treatment adherence, age, gender, nutritional status, disease states, and genetic polymorphisms that may influence risk of adverse events and treatment resistance (Figure 50-1). Drug-drug interactions are an additional factor that influences how patients react to drugs. The importance of these interactions depends heavily on the clinical context. In many cases, the practical impact of drug-drug interactions is likely to be very small compared with other factors that affect treatment response, drug levels, and toxicity. It is reasonable therefore to focus special attention on the contexts in which drug-drug interactions are most likely to be clinically problematic.
CLASSIFICATION
Drug-drug interactions may be described as pharmacokinetic, pharmacodynamic, idiosyncratic, or mixed, depending on the presumed mechanism underlying the interaction (Table 50-1). Pharmacokinetic interactions are those that involve a change in the plasma level or tissue distribution (or both) of one drug by virtue of co-administration of another drug. These interactions occur due to effects at one or more of the four pharmacokinetic processes by which drugs are acted on by the body: absorption, distribution, metabolism, and excretion. Because of the importance of these factors, particularly metabolism, in drug-drug interactions, a more detailed de-scription of pharmacokinetic processes follows. An example of a pharmacokinetic drug-drug interaction is the inhibition of the metabolism of lamotrigine by valproic acid,12 thereby raising lamotrigine levels and increasing the risk of potentially serious adverse events, including hypersensitivity reactions (such as Stevens-Johnson syndrome). In contrast, pharmacodynamic interactions are those that involve a known pharmacological effect at biologically active (receptor) sites. These interactions occur due to effects on the mechanisms through which the body is acted on by drugs and do not involve a change in drug levels. An example of a pharmacodynamic drug-drug interaction is the interference of the antiparkinsonian effects of a dopamine receptor agonist (such as pramipexole) by a dopamine receptor antagonist (such as risperidone). Mixed interactions are those that are believed to involve both pharmacological and pharmacodynamic effects. The apparent enhancement of antidepressant response observed on the combination of fluoxetine and desipramine, for example, may be attributed to the pharmacokinetic elevation of desipramine blood levels due to inhibition of its metabolism by fluoxetine, as well as to putative synergistic pharmacodynamic actions of these two agents at serotonergic and noradrenergic synapses. Finally, idiosyncratic interactions are those that occur sporadically in small numbers of patients in ways that are not yet predicted by the known pharmacokinetic and pharmacodynamic properties of the drugs involved.
Table 50-1 Classification of Drug-Drug Interactions
| Pharmacokinetic |
| Alteration in blood level or tissue concentration (or both) resulting from interactions involving drug absorption, distribution, metabolism, or excretion |
| Pharmacodynamic |
| Alteration in pharmacological effect resulting from interactions at the same or interrelated biologically active (receptor) sites |
| Mixed |
| Alterations in blood levels and pharmacological effects due to pharmacokinetic and pharmacodynamic interactions |
| Idiosyncractic |
| Sporadic interactions among drugs not accounted for by their currently known pharmacokinetic or pharmacodynamic properties |
PHARMACOKINETICS
As described earlier, pharmacokinetic processes refer to absorption, distribution, metabolism, and excretion, factors that determine plasma levels and tissue concentrations of a drug.2,11,13 Pharmacokinetics refers to the mathematical analysis of these processes. Advances in analytic chemistry and computer methods of pharmacokinetic modeling and a growing understanding of the molecular pharmacology of the liver enzymes responsible for metabolism of most psychotropic medications have furnished increasingly sophisticated insights into the disposition and interaction of administered drugs. Although pharmacokinetics refers to only one of the two broad mechanisms by which drugs interact, pharmacokinetic interactions involve all classes of psychotropic and nonpsychotropic medications. An overview of pharmacokinetic processes is a helpful prelude to a discussion of drug-drug interactions by psychotropic drug class.
Distribution
Drugs distribute to tissues through the systemic circulation. The amount of drug ultimately reaching receptor sites in tissues is determined by a variety of factors, including the concentration of free (unbound) drug in plasma, regional blood flow, and physiochemical properties of drug (e.g., lipophilicity or structural characteristics). For entrance into the central nervous system (CNS), penetration across the blood-brain barrier is required. Fat-soluble drugs (such as benzodiazepines, neuroleptics, and cyclic antidepressants) distribute more widely in the body than water-soluble drugs (such as lithium), which distribute through a smaller volume of distribution. Changes with age, typically including an increase in the ratio of body fat to lean body mass therefore result in a net greater volume of lipophilic drug distribution and potentially greater accumulation of drug in adipose tissue in older than in younger patients.
An emerging understanding of the drug transport proteins, of which P-glycoproteins are the best characterized, indicates a crucial role in regulating permeability of intestinal epithelia, lymphocytes, renal tubules, the biliary tract, and the blood-brain barrier. These transport proteins are thought to account for the development of certain forms of drug resistance and tolerance, but are increasingly seen as likely also to mediate clinically important drug interactions.2 Little is known yet about their relevance to drug interactions involving psychiatric medications; the capacity of St. John’s wort to lower blood levels of several critical medications (including cyclosporine and indinavir) is hypothesized to be related, at least in part, to an effect of the botanical agent on this transport system.14
Metabolism
Metabolism is the best-characterized mechanism of all of the pharmacokinetic processes implicated in known drug-drug interactions. Metabolism refers to the biotransformation of a drug to another form, a process that is usually enzyme-mediated and that results in a metabolite that may or may not be pharmacologically active and may or may not be subject to further biotransformations before eventual excretion. Most drugs undergo several types of biotransformation, and many psychotropic drug interactions of clinical significance are based on interference with this process. A growing understanding of hepatic enzymes, and especially the rapidly emerging characterization of the cytochrome P450 isoenzymes and other enzyme systems including the uridine-diphosphate glucuronosyltransferases (UGTs), flavin-containing monooxygenases (FMOs), methyltransferases, and sulfotransferases, has significantly advanced a rational understanding and prediction of drug interactions and individual variation in drug responses.2,15
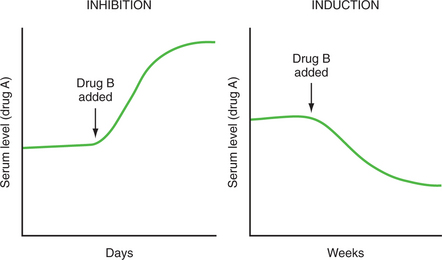
The synthesis or activity of hepatic microsomal enzymes is affected by metabolic inhibitors and inducers, as well as by distinct genetic polymorphisms (stably inherited traits). Table 50-2 lists enzyme inducers and inhibitors common in clinical settings. These should serve as red flags that beckon further scrutiny for potential drug-drug interactions when they are found on a patient’s medication list. Imagine two drugs, drug A and drug B, which are both associated with a metabolic enzyme. Drug B may be an inhibitor or an inducer of that enzyme. Drug A may be normally metabolized by that enzyme and would therefore be called a substrate. If drug B is an inhibitor with respect to the metabolic enzyme, it will impede the metabolism of a concurrently administered substrate (drug A), thereby producing a rise in the plasma levels of that substrate. If drug B is an inducer of that enzyme, it will enhance the metabolism of the substrate (drug A), resulting in a decline in the plasma levels of that substrate (Figure 50-2). In some circumstances an inhibitor (such as grapefruit juice) or inducer (e.g., a cruciferous vegetable, such as brussels sprouts) may not be a drug but rather another ingested substance. Moreover, in some circumstances a drug is not only a substrate of an enzyme but can also inhibit the metabolism of other substrates relying on that enzyme, in which case it is considered an inhibitor as well as a substrate. Although inhibition is usually immediate, occurring by one or more of a variety of mechanisms (including competitive inhibition or inactivation of the enzyme), induction, which requires enhanced synthesis of the metabolic enzyme, is typically a more gradual process. A fall in plasma levels of a substrate may not be apparent for days to weeks following introduction of the inducer. This is particularly important when a patient’s care is being transferred to another setting where clinical deterioration may be the first sign that drug levels have declined. Reciprocally, an elevation in plasma drug concentrations could reflect the previous discontinuation of an inducing factor (e.g., cigarette smoking or carbamazepine) just as it could reflect the more recent introduction of an inhibitor (e.g., fluoxetine or valproic acid).
Table 50-2 Commonly Used Drugs and Substances That Inhibit or Induce Hepatic Metabolism of Other Medications
| Inhibitors | Inducers |
|---|---|
| Antifungals (ketoconazole, miconazole, itraconazole) | Barbiturates (e.g., phenobarbital, secobarbital) |
| Macrolide antibiotics (erythromycin, clarithromycin, triacetyloleandomycin) | Carbamazepine |
| Oxcarbazepine | |
| Fluoroquinolones (e.g., ciprofloxacin) | Phenytoin |
| Isoniazid | Rifampin |
| Antiretrovirals | Primidone |
| Antimalarials (chloroquine) | Cigarettes |
| Selective serotonin reuptake inhibitors (fluoxetine, fluvoxamine, paroxetine, sertraline) | Ethanol (chronic) |
| Cruciferous vegetables | |
| Nefazodone | Charbroiled meats |
| β-Blockers (lipophilic) (e.g., propranolol, metoprolol, pindolol) | St. John’s wort |
| Quinidine | |
| Valproate | |
| Cimetidine | |
| Calcium channel blockers (e.g., diltiazem) | |
| Grapefruit juice | |
| Ethanol (acute) |
Although the cytochrome P450 isoenzymes represent only one of the numerous enzyme systems responsible for drug metabolism, they are responsible for metabolizing, at least in part, over 80% of all prescribed drugs. In addition, growing awareness in the 1990s about the capacity of many of the newer antidepressants to inhibit cytochrome P450 isoenzymes fueled great interest in the pattern of interaction of psychotropic and other drugs with these enzymes in the understanding and prediction of drug-drug interactions. The cytochrome P450 isoenzymes represent a family of more than 30 related heme-containing enzymes, largely located in the endoplasmic reticulum of hepatocytes (but also present elsewhere, including the gut and brain), which mediate oxidative metabolism of a wide variety of drugs, as well as endogenous substances (including prostaglandins, fatty acids, and steroids). The majority of antidepressant and antipsychotic drugs is metabolized by, or inhibits, one or more of these isoenzymes. Table 50-3 summarizes the interactions of psychiatric and nonpsychiatric drugs with a subset of isoenzymes that have been increasingly well characterized (1A2, 2C subfamily, 2D6, and 3A4). Further information about the relevance of these and other interactions is highlighted in a later section of this chapter in which clinically important drug-drug interactions are reviewed.
Table 50-3 Selected Cytochrome P450 Isoenzyme Substrates, Inhibitors, and Inducers
| 1A2 | Substrates | Acetaminophen, aminophylline, caffeine, clozapine, cyclobenzaprine, estradiol, fluvoxamine, haloperidol, mirtazapine, ondansetron, olanzapine, phenacetin, procarcinogens, propranolol, ramelteon, riluzole, ropinirole, tacrine, tertiary amine tricyclic antidepressants (TCAs), theophylline, verapamil, warfarin, zileuton, zolmitriptan |
| Inhibitors | Amiodarone, cimetidine, fluoroquinolones, fluvoxamine, grapefruit juice, methoxsalen, ticlopidine | |
| Inducers | Charbroiled meats, tobacco (cigarette smoking), cruciferous vegetables, modafinil, omeprazole | |
| 2C | Substrates | Barbiturates, diazepam, fluvastatin, glipizide, glyburide, irbesartan, losartan, mephenytoin, NSAIDs, nelfinavir, phenytoin, primidone, propranolol, proguanil, proton pump inhibitors, rosiglitazone, tamoxifen, tertiary TCAs, THC, tolbutamide, R-warfarin, S-warfarin |
| Inhibitors | Fluoxetine, fluvoxamine, ketoconazole, modafinil, omeprazole, oxcarbazepine, sertraline | |
| Inducers | Carbamazepine, norethindrone, prednisone, rifampin, secobarbital | |
| 2D6 | Substrates | Aripiprazole, atomoxetine, beta-blockers (lipophilic), codeine, debrisoquine, dextromethorphan, diltiazem, donepezil, duloxetine, encainide, flecainide, haloperidol, hydroxycodone, lidocaine, metoclopramide, mexiletine, mCPP, nifedipine, ondansetron, phenothiazines (e.g., thioridazine, perphenazine), promethazine, propafenone, risperidone, SSRIs, tamoxifen, TCAs, tramadol, trazodone, venlafaxine |
| Inhibitors | Amiodarone, antimalarials, bupropion, cimetidine, citalopram, duloxetine, escitalopram, fluoxetine, methadone, metoclopramide, moclobemide, paroxetine, phenothiazines, protease inhibitors (ritonavir), quinidine, sertraline, terbinafine, TCAs, yohimbine | |
| Inducers | Dexamethasone, rifampin | |
| 3A3/4 | Substrates | Alfentanil, alprazolam, amiodarone, amprenavir, aripiprazole, bromocriptine, buspirone, cafergot, calcium channel blockers, caffeine, carbamazepine, cisapride, clozapine, cyclosporine, dapsone, diazepam, disopyramide, efavirenz, estradiol, fentanyl, indinavir, HMG-CoA reductase inhibitors (lovastatin, simvastatin), lidocaine, loratadine, methadone, midazolam, nimodipine, pimozide, prednisone, progesterone, propafenone, quetiapine, quinidine, ritonavir, sildenafil, tacrolimus, testosterone, tertiary amine TCAs, triazolam, vinblastine, warfarin, zolpidem, zaleplon, ziprasidone |
| Inhibitors | Antifungals, calcium channel blockers, cimetidine, efavirenz, indinavir, fluvoxamine, fluoxetine (norfluoxetine), grapefruit juice, macrolide antibiotics, mibefradil, nefazodone, ritonavir, verapamil, voriconazole | |
| Inducers | Carbamazepine, glucocorticoids, modafinil, oxcarbazepine, phenobarbital, phenytoin, pioglitazone, rifabutin, rifampin, ritonavir, St. John’s wort, troglitazone |
HMG-CoA, Hydroxymethylglutaryl coenzyme A; mCPP, m-chlorophenylpiperazine; NSAIDs, nonsteroidal antiinflammatory drugs; SSRIs, selective serotonin reuptake inhibitors; THC, tetrahydrocannabinol.
In addition to being influenced by pharmacological inducers or inhibitors, a patient’s metabolic status is also under genetic control. Knowledge continues to evolve concerning genetic polymorphisms that affect drug metabolism. Within the group of cytochrome P450 isoenzymes, there appears to be a polymodal distribution of metabolic activity in the population with respect to certain isoenzymes (including 2C19 and 2D6). Most individuals are normal (“extensive”) metabolizers with respect to the activity of these isoenzymes. A smaller number are “poor metabolizers” with deficient activity of the isoenzyme. Probably very much smaller numbers are ultra-rapid metabolizers (who have more than normal activity of the enzyme) and intermediate metabolizers (who fall between extensive and poor metabolizers). Individuals who are poor metabolizers with respect to a particular cytochrome P450 isoenzyme are expected to have higher plasma concentrations of a drug that is metabolized by that isoenzyme, thereby potentially being more sensitive to or requiring lower doses of that drug than a patient with normal activity of that enzyme. They may also have higher than usual plasma levels of metabolites of the drug that are produced through other metabolic pathways that are not altered by the polymorphism, thereby potentially incurring pharmacological activity or adverse effects related to these alternative metabolites. Poor metabolizers are relatively impervious to drug interactions involving inhibition of the particular isoenzyme system for which they are already deficient. Studies on genetic polymorphisms affecting the cytochrome P450 system suggest ethnic differences.16 Approximately 15% to 20% of Asian Americans and African Americans appear to be poor metabolizers with respect to P450 2C19 compared with 3% to 5% of Caucasians. Conversely, the proportion of frankly poor metabolizers with respect to P450 2D6 appears to be higher among Caucasians (approximately 5% to 10%) than among Asian Americans and African Americans (approximately 1% to 3%). Current understanding of the clinical relevance of genetic polymorphisms in drug therapy in psychiatry remains rudimentary. Commercial genotyping tests for polymorphisms of potential relevance to drug-drug metabolism are increasingly available. Further systematic study of their relevance to the understanding and prediction of drug response is needed before such testing can be meaningfully incorporated into routine psychopharmacological practice.
Excretion
Because most antidepressant, anxiolytic, and antipsychotic medications are largely eliminated by hepatic metabolism, factors that affect renal excretion (glomerular filtration, tubular reabsorption, and active tubular secretion) are generally far less important to the pharmacokinetics of these drugs than to lithium, for which such factors may have clinically significant consequences. Conditions resulting in sodium deficiency (e.g., dehydration, sodium restriction, and use of thiazide diuretics) are likely to result in increased proximal tubular reabsorption of lithium, resulting in increased lithium levels and potential toxicity. Lithium levels and clinical status must be monitored especially closely in the setting of vomiting, diarrhea, excessive evaporative losses, or polyuria. Factors, such as aging, that are associated with reduced renal blood flow and glomerular filtration rate (GFR) also reduce lithium excretion. For this reason, as well as for their reduced volume of distribution for lithium because of relative loss of total body water with aging, elderly patients typically require lower lithium doses than younger patients, and a low starting dose (i.e., 150 to 300 mg/day) is often prudent. Apparently separate from pharmacokinetic effects, however, elderly patients may also be more sensitive to the neurotoxic effects of lithium even at low therapeutic levels. Factors associated with an increased GFR, particularly pregnancy, may produce an increase in lithium clearance and a fall in lithium levels.
ANTIPSYCHOTICS
The antipsychotic or neuroleptic drugs include the phenothiazines (e.g., chlorpromazine, fluphenazine, perphenazine, thioridazine, and trifluoperazine), butyrophenones (haloperidol), thioxanthenes (thiothixene), indolones (molindone), diphenylbutylpiperidines (pimozide), dibenzodiazepines (loxapine), and the newer atypical agents (clozapine, olanzapine, risperidone, quetiapine, ziprasidone, and aripiprazole).17 As a class, they are generally rapidly, if erratically, absorbed from the gastrointestinal tract after oral administration (peak plasma concentrations ranging from 30 minutes to 6 hours). They are highly lipophilic and distribute rapidly to body tissues with a large apparent volume of distribution. Protein-binding in the circulation ranges from approximately 90% to 98% except for molindone and quetiapine, which are only moderately protein-bound. The antipsychotics generally undergo substantial first-pass hepatic metabolism (primarily oxidation and conjugation reactions), reducing their systemic bioavailability when given orally compared with intramuscular (IM) administration, the fractional absorption of which nearly approximates that of IV administration. Most of the individual antipsychotics have several pharmacologically active and yet other inactive metabolites. Because of their propensity to sequester in body compartments, the elimination half-life of antipsychotics is quite variable, generally ranging from approximately 20 to 40 hours. For butyrophenones, however, elimination pharmacokinetics appear to be especially complex, and the disappearance of drug from the systemic circulation, and even more so from brain, may take much longer, as it does for the newer agent, aripiprazole (and its active metabolite, dehydro-aripirazole), whose half-life may exceed 90 hours.





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


