Parkinson’s Disease
PD is due to a degeneration of neurons in the dopaminergic nigrostriatal pathway. It is the second most common movement disorder behind essential tremor (ET), affecting about 1% of the population over the age of 50. The prevalence increases exponentially between the ages of 65 and 90, and reaches 3% of the population over 65. Cardinal manifestations include bradykinesia, rigidity, tremor, an expressionless face, and postural instability. Asymmetry is characteristic. The disease often begins asymmetrically; the signs may be so lateralized as to warrant the designation of hemi-PD, and some asymmetry usually persists even when the disease is well established. The major manifestations vary from case to case. In some, tremor is the outstanding symptom and in others the rigidity, the bradykinesia, or the loss of associated movements. Asymmetric rest tremor is the most common presentation of PD, occurring in about 75% of patients. Akinetic, tremor, and postural instability subtypes have been recognized.
PD causes marked hypertonia, or rigidity, which principally affects the axial muscles and the proximal and flexor groups of the extremities, causing an increased tone to passive movement. The rigidity has a rhythmic quality referred to as cogwheel rigidity (Negro’s sign), presumably due to the superimposition of the tremor. Cogwheeling may be brought out as the examiner passively moves an elbow or wrist by having the patient grit the teeth, look at the ceiling, or use the opposite hand to make a fist, trace circles in the air, or imitate throwing a ball. The rigidity is present evenly throughout the range of movement, without the ebb at the extremes of the range that occurs in spasticity.
In PD, there is a paucity of movement and a slowing of movements. Strictly speaking, akinesia means an absence of movement; bradykinesia, a slowness of movement; and hypokinesia, a decreased amount or amplitude of movement, but the term bradykinesia is often used to encompass all three. Bradykinesia is not due simply to the rigidity and may have an independent pathophysiologic basis. There is loss of associated and automatic movements, with masking of the face, infrequent smiling and blinking, and loss of swinging of the arms in walking (Figure 30.1). Normal fidgeting and adventitial movements are decreased or absent. Because of the rigidity and bradykinesia, strength may seem to be decreased, but (although an alternate name for PD is paralysis agitans, and Parkinson referred to the disorder as “shaking palsy”) there is no true loss of power such as is seen in corticospinal lesions. The rigidity and bradykinesia involve movements and not muscles or muscle groups, and do make locomotion and motor activity slow and difficult. Under acute emotional stress, the extremities can often be used rapidly and effectively, as when an otherwise immobile patient escapes from a fire (kinesia paradoxica).
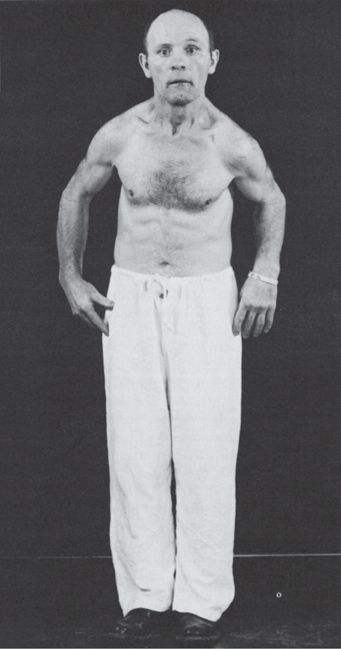
FIGURE 30.1 A patient with Parkinson’s disease, showing rigidity, masked facies, and typical posture.
The tremor of PD is a coarse “pill-rolling” movement, so named because of its resemblance to the motion pharmacists of a bygone era used to make pills. The tremor is fairly rhythmic, gross, from 2 to 6 Hz, and may involve the hands, feet, jaw, tongue, lips, and pharynx, but not the head. It is typically a resting tremor that lessens during voluntary movement and disappears in sleep. Parkinson said the tremor was present “whilst the limb is at rest and unemployed.” The tremor fluctuates, increasing in amplitude but not rate when the patient becomes excited. The tremor often is more apparent when the patient is walking. Some patients may also have a low-amplitude 7 to 8 Hz tremor during voluntary movement that is suppressed by relaxation.
Patients with PD have poor balance, a tendency to fall, and difficulty walking. Gait and balance are not prominently affected in most patients with early PD, and significant postural instability early in the course suggests an alternate diagnosis, such as progressive supranuclear palsy (PSP) (see below). The gait abnormality is stereotypical: slow and shuffling with a reduced stride length, sometimes markedly so; a stooped flexed posture of the body and extremities (simian posture); reduced arm swing; and a tendency to turn “en-bloc.” Head drop and camptocormia may occur. Lower-half (lower-body, arteriosclerotic) parkinsonism causes gait difficulty out of proportion to other manifestations (Chapter 44). Impaired postural reflexes lead to a tendency to fall forward (propulsion), which the patient tries to avoid by walking with increasing speed but with very short steps, the festinating gait. Falls are common. If a patient, standing upright, is gently pushed either backward or forward, she cannot maintain balance and will fall in the direction pushed. When a patient, seated in a chair, is suddenly tilted backward, there is absence of the normal reflex leg extension to counteract the loss of balance (Souques’ leg sign). Facial immobility and lack of expressiveness is a common feature of PD (hypomimia, masked face). A decreased rate of blinking (5 to 10 per minute rather than the normal 12 to 20), accompanied by slight eyelid retraction (Stellwag’s sign, also seen in thyroid eye disease), causes patients to have a staring expression (reptilian stare). Abnormalities of phonation and articulation are common (bradylalia). The voice is typically soft, breathy, monotonous, and tremulous. Lack of movement of the lips and tongue causes articulatory imprecision. Temporarily overcoming vocal bradykinesia, words pour out in a short rush of rapid speech. Patients with PD tend to be soft, fast, mumbly talkers. They are often unable to speak loudly or shout. There is also slowness of chewing and swallowing, and the decreased swallowing is largely responsible for the drooling that sometimes occurs.
The freezing phenomenon is common in PD. In the midst of a motor act, the patient will suddenly freeze in place, unable to move for a short time because of simultaneous activation of agonists and antagonists. Freezing may occur when first starting to walk (start-hesitation), when turning (turn-hesitation), when approaching an obstacle, and even when talking or eating. Associated involvement of midbrain structures may cause changes in ocular movements, including fixation instability, hypometric saccades, convergence insufficiency, and impaired upgaze. Oculogyric crisis, forced involuntary eye deviation, usually upward, is a feature of postencephalitic PD and can occur in drug-induced parkinsonism, but it does not happen in idiopathic PD. Other common manifestations include hyperhidrosis, greasy seborrhea, micrographia, somnolence, difficulty turning over in bed, blepharospasm, and apraxia of eyelid opening.
In PD, there is no atrophy, fasciculations, reflex changes, or pathologic reflexes of the type seen in corticospinal tract disorders (see Table 22.1). Reflex changes may occur if there is associated corticospinal tract involvement, but this does not occur in idiopathic PD. Even when the extrapyramidal signs are asymmetric, the reflexes remain normal and equal. Fragments of dystonia sometimes occur. A “striatal toe” (dystonic toe) is an apparent extensor plantar response, without fanning of the toes that occurs in isolation, without other signs suggesting corticospinal tract dysfunction, in patients with extrapyramidal disorders such as PD. The extended toe may occur as part of a foot dystonia that includes ankle inversion, arching of the sole and flexion of the other toes (striatal foot). The parkinsonian hand is slightly extended at the wrist and flexed at the metacarpophalangeal joints with the fingers extended and adducted. There is often exaggeration of the orbicularis oculi and orbicularis oris reflexes. Myerson’s sign (glabellar tap reflex) is blinking of the eyes on tapping over the glabella. In PD, the patient is unable to inhibit the response and will continue to blink over and over; normals do not continue to blink with repetitive tapping (see Chapter 16). Patients with PD may exhibit a variety of nonmotor manifestations, such as anosmia, seborrhea, constipation, REM sleep behavior disorder, depression, and dementia. Nonmotor manifestations may occur before motor symptoms develop.
In the early stages of PD, typical signs are often subtle and patients may present complaining only of stiffness, impaired handwriting (especially micro-graphia), or difficulty getting about. Stiffness and myalgic pains may suggest a diagnosis of arthritis, polymyalgia, or fibromyalgia. The facial masking and bradykinesia often lead to a misdiagnosis of depression. Early PD may also be mistaken for the effects of aging. Rest tremor, a flexed posture, and mild cogwheel rigidity provide important clues to the possibility of early parkinsonism in such patients. The clinical diagnosis of PD at the time of initial presentation is occasionally wrong. The pattern of disease progression and the response to medication are important additional factors in determining whether the patient suffers from PD or one of the other akinetic-rigid syndromes.
Advancing disease is characterized by increasing gait difficulty and worsening of tremor and bradykinesia. Other major problems in advanced PD include motor fluctuations related to levodopa therapy, behavioral changes, cognitive impairment, depression, hallucinations, impotence, dysphagia, speech difficulty, intractable drooling, and sleep impairment. The impairment of cognition in PD is extremely variable, ranging from minimal involvement to profound dementia. Some degree of cognitive blunting may occur in 20% to 40% of patients. Severe dementia is rare; when present, especially early in the course, the possibility of dementia with Lewy bodies should be considered. Depression occurs in approximately one-third of patients, and psychosis and hallucinations in about one-fourth of patients. The hallucinations are primarily visual and can be very chronic and persistent. Early, prominent, and nonvisual hallucinations raise the possibility of dementia with Lewy bodies. The absence of tremor in the early stages may be associated with a greater likelihood of dementia in the late stages and also may suggest a possible parkinsonplus syndrome. Advanced age at onset, severe depression, dementia, and an akinetic-rigid presentation are risk factors for rapid disease progression. The tremor may begin to abate in the very late stages.
PD usually affects older patients, the mean age as onset is around 60 years but some cases began relatively early in life. The nosology describing these patients is inconsistent. Juvenile Parkinson’s disease (JPD) has its onset before 20, and young onset Parkinson’s disease (YOPD), between 20 and 50, but the terms are sometimes used interchangeably. JPD was first described by Ramsay Hunt and is one of the several “Ramsay Hunt syndromes.” YOPD is probably the same entity as idiopathic PD, with a younger age of onset. In a series of 953 individuals with YOPD, 17% were found to have a genetic etiology. In contrast, JPD is a heterogeneous group of disorders. Many patients with JPD have a genetic disorder due to a parkin gene mutation; 16 parkin mutations are currently recognized. Patients with parkin mutations may have atypical clinical features such as dystonia at onset and marked diurnal symptom fluctuation; the disorder may bear some relationship to other forms of dystonia. Parkin mutations can also cause a disorder clinically similar to idiopathic sporadic PD, but without Lewy bodies. YOPD tends to have more gradual progression of parkinsonian signs and symptoms and earlier treatment-related complications. Susceptibility to developing late-onset PD has been associated with polymorphisms or mutations in several genes. Mutations of the LRRK2 gene are the commonest cause of familial PD, at least six mutations have been identified, and are responsible for approximately 1% of typical sporadic cases of the disease.
The diagnosis of PD is predominantly clinical, and differential diagnosis essentially is between other conditions causing tremor, of which ET is the commonest, and other akinetic-rigid syndromes. Clinical features that favor PD include prominent rest tremor, asymmetric signs, preservation of balance and postural reflexes in the early stages of the disease, and a good response to levodopa replacement therapy. The other degenerative disorders with parkinsonian features typically produce other neurologic signs, such as gaze limitation, cerebellar signs, pyramidal signs, severe dementia, apraxia and other parietal lobe signs, or dysautonomia, although these other manifestations may not be apparent early in the course. In the past, a diagnosis of PD required two of three parkinsonian features (tremor, rigidity, bradykinesia). Using these criteria resulted in a 24% error rate based on pathologic studies. Using revised criteria (the UK brain bank criteria) of rest tremor, asymmetry, and a good response to levodopa improved accuracy and resulted in pathologic confirmation of the diagnosis in 99% of cases determined by clinicopathologic correlation studies. The most common condition confused with PD is PSP (see below).
Certain drugs can induce a reversible condition that mimics PD. The most common agents that cause drug-induced parkinsonism are antipsychotics, especially the high-potency piperazine compounds such as haloperidol. The atypical neuroleptics are as potent in their antipsychotic effects as traditional compounds but less likely to induce parkinsonism. Drug-induced parkinsonism can mimic PD closely, even to the point of causing asymmetric signs. Although dopamine receptor-blocking agents, especially those that block the D2 receptor, are the most common offenders, other agents can induce parkinsonism. Some patients are much more prone to develop extrapyramidal side effects than others, but most individuals will eventually develop parkinsonism if treated with high doses. Metoclopramide is a dopamine blocker most often used for gastrointestinal disease; it may have extrapyramidal side effects.
In the presence of typical clinical signs and symptoms and the typical age of onset, extensive workup is not required. Imaging studies are usually normal. A thorough medication history, complete neurologic examination to look for nonextrapyramidal abnormalities, and screening for orthostatic hypotension are useful. Certain features suggest an alternative diagnosis, such as prominent dementia and hallucinations in the patient suffering from dementia with Lewy bodies, prominent dysautonomia in the patient with multisystem atrophy (MSA), dysarthria, and early age of onset in Wilson’s disease.
Pathologically, the disease is characterized by depigmentation and cystic degeneration of the pars compacta of the substantia nigra with cell loss and the presence of intracytoplasmic Lewy bodies in surviving neurons. A major chemical constituent of the Lewy body is alpha-synuclein, a synaptic protein. Abnormal aggregation of alpha-synuclein has been advocated in the pathogenesis of PD, dementia with Lewy bodies, and multiple system atrophy, and these conditions have been grouped together as synucleinopathies. In PD, there is depletion of dopamine; replacement by its precursor, levodopa, has been well recognized as effective therapy since the landmark studies of the 1960s. The etiology of PD remains unknown, but the basis is likely multifactorial, possibly involving hereditary factors, environmental influences that may selectively affect dopaminergic nigral cells, and free radical toxicity. Mutations in the genes coding for alpha-synuclein have been identified in patients with autosomal dominant (AD) PD and mutations in the parkin gene in autosomal recessive PD. Abnormalities of mitochondrial DNA have been implicated in other cases. Age-dependent penetrance may be a complicating factor in discerning any genetic influence. Mitochondrial dysfunction is suggested by abnormalities of complex I of the mitochondrial electron transport change in PD, and the ability of the known parkinsonogenic toxin MPTP (1-N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) to affect complex I. There is evidence that complex I inhibition may be the central cause of sporadic PD and that abnormalities of complex I underlie the alpha-synuclein aggregation. The metabolic reactions synthesizing dopamine also produce free radicals. The substantia nigra of PD patients contains increased levels of iron, which promote free radical formation and decreased levels of the free radical inhibitor glutathione. This may in part explain the susceptibility of nigral neurons to oxidative stress.
The pathophysiology of parkinsonism is complex. Dopamine deficiency ultimately results in an increased output from the internal segment of the globus pallidus and subthalamic nuclei, which results in excessive inhibition of the thalamus and suppression of the cortical motor system (see Chapter 26). Pharmacologic treatment modalities include anticholinergic drugs, dopamine releasing agents, dopamine agonists that directly stimulate the dopamine receptor in the striatum, catechol-O-methyl transferase inhibitors, and levodopa. Dopamine and acetylcholine are in balance in the striatum, and anticholinergics increase the effect of dopamine by influencing this balance. These agents were the earliest treatment available for PD. The discovery of L-dopa as a treatment for PD was a major medical advance. The story of that discovery is told in British neurologist Oliver Sacks’ book, Awakenings, and its motion picture adaptation. Over 90% of patients with PD respond very well to the initiation of levodopa replacement. The absence of a good initial response suggests the possibility of another diagnosis, although some of the mimickers of PD, especially MSA, may also have a good initial response. Several surgical approaches have been used, including “brain transplants” with transplantation of adrenal medullary cells or fetal mesencephalic tissue into the striatum, as well as thalamotomy, pallidotomy, and deep brain stimulation. Surgical approaches have revolutionalized the management of PD.
Some of the other conditions important in the differential diagnosis of PD include multiple system atrophy, PSP, corticobasal degeneration (CBD), and diffuse Lewy body disease.
Multisystem Atrophy
MSA produces degeneration involving the basal ganglia, cerebellum, anterior horn cells, cerebral cortex, and brainstem in varying combinations, and usually includes parkinsonian features. Patients with MSA may have elements of cerebellar ataxia, dementia, amyotrophy, parkinsonism, corticospinal tract dysfunction, dysautonomia, and urinary dysfunction.
There are three subtypes: MSA-P (parkinsonian), MSA-C (cerebellar), and MSA-A (autonomic). In MSA-P (formerly striatonigral degeneration), the primary manifestation is parkinsonism; it accounts for about 80% of the cases of MSA. About 10% of patients presenting with parkinsonism will evolve into MSA-P. In MSA-C (formerly the sporadic form of olivopontocerebellar atrophy), the primary manifestations are cerebellar. In MSA-A (formerly Shy-Drager syndrome), the primary manifestation is dysautonomia, particularly postural hypotension. Suspicion of MSA should arise in the patient with atypical parkinsonism in conjunction with cerebellar signs and/or early and prominent autonomic dysfunction, usually orthostatic hypotension. There is some overlap of pathologic findings in the different forms of MSA. Recently discovered glial cytoplasmic inclusions containing a-synuclein confirm these three disorders are different forms of the same disease. There may be a cytoskeletal abnormality in glial cells that leads to neuronal degeneration. A lack of levodopa responsiveness has been used as another distinguishing characteristic of MSA, but a sizeable minority of patients does respond initially. The dysautonomia is due to degeneration of the neurons in the intermediolateral gray column of the thoracic and lumbar spinal cord. The intermediolateral gray column degeneration can occur without any other abnormalities, causing a syndrome of idiopathic orthostatic hypotension. Impaired peripheral vasomotor control in MSA may cause cold, dusky, purplish discoloration of the digits (cold hands sign). Dysautonomia can also occur in association with degeneration of other parts of the nervous system.
Parkinsonism is the most frequent motor manifestation of MSA. MSA-P has been associated with relative symmetry of findings, urinary incontinence, frequent falls, an absence of tremor, and the frequent presence of pyramidal or cerebellar signs. MSA-A consists of a combination of parkinsonian signs and symptoms coupled with severe dysautonomia, most often manifest as orthostatic hypotension. The primary manifestations of MSA-C are ataxia and brainstem dysfunction. Approximately one quarter of patients with MSA-C will develop parkinsonian features within 5 years; such evolution carries a poor prognosis for survival.
Progressive Supranuclear Palsy
In PSP (Steele-Richardson-Olszewski syndrome, Richardson’s syndrome), degenerative changes in the rostral brainstem and thalamus result in impairment first of downgaze, then of upgaze, and eventually in global gaze paresis. Three subtypes are currently recognized: classic Richardson’s syndrome (PSP-RS), PSP parkinsonism (PSP-P) and pure akinesia with gait freezing. In PSP-P, the clinical picture resembles PD early in the course. In PSP-RS, the gaze abnormalities are accompanied by parkinsonian signs, frontal lobe type dementia, postural instability, pseudobulbar palsy, and a pronounced tendency to extensor axial rigidity, especially involving the neck muscles, and sometimes causing frank retrocollis. Gait difficulty is prominent, and a tendency to fall is an early and conspicuous feature. The falls are often backward because of the increased extensor tone and postural instability. There is particular difficulty walking down stairs because of the combination of retrocollis and impaired downgaze. Tremor is not usually pronounced, and the disease does not respond significantly to levodopa. The gaze abnormalities at first involve voluntary, vertical, saccadic gaze, sparing reflex eye movements; the name of the disease refers to this supranuclear ocular motility disturbance. The disordered motility may progress to complete ophthalmoplegia terminally. Facial dystonia may contort the face, particularly the forehead muscles, into a characteristic expression of “perpetual surprise” or astonishment with raised eyebrows, lid retraction, and reduced blinking (procerus sign). The facial expression is markedly different from the hypomimia and masking of PD. The characteristic ocular motility disturbance may not be present early in the course, and, in rare instances, never appears. The “applause sign” is an inability to stop clapping after being asked to clap three times. It was initially touted as a way to distinguish PSP from PD and frontotemporal dementia. Later studies found the applause sign in cortical dementia and suggested that it is a nonspecific sign of frontal lobe dysfunction. On magnetic resonance imaging (MRI), a characteristic atrophy of the midbrain with relative preservation of the pons (the “hummingbird sign” on midsagittal images) may be seen (Figure 30.2). MRI measurement of the midbrain/pons ratio and combined measurements, such as midbrain area/pons area and especially the magnetic resonance parkinsonism index are very useful in distinguishing PSP from other parkinsonian syndromes.
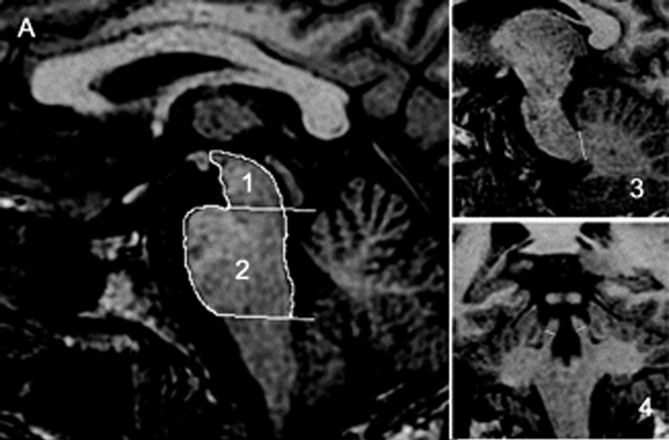
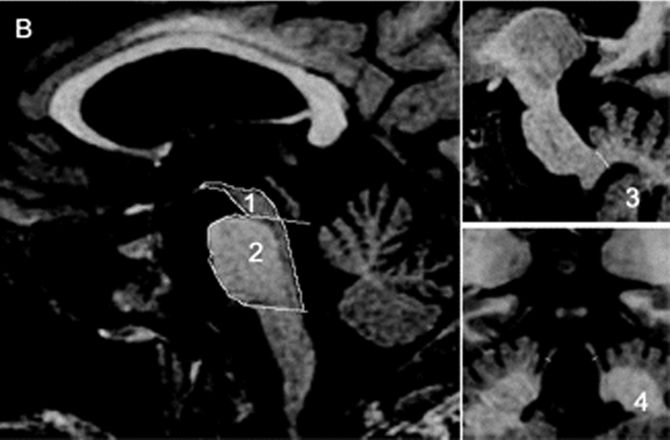
FIGURE 30.2 Sagittal and coronal T1-weighted volumetric spoiled gradient-echo MR images of (A) a patient with clinically unclassifiable parkinsonism with normal magnetic resonance parkinsonism index (MRPI) and (B) a patient with progressive supranuclear palsy (PSP). There is marked atrophy of both midbrain and superior cerebellar peduncle in the patient with PSP. (Reprinted from Morelli M, Arabia G, Novellino F, et al. MRI measurements predict PSP in unclassifiable parkinsonisms: a cohort study. Neurology 2011;77:1042–1047, with permission.)
Corticobasal Syndrome
In CBD (cortical-basal ganglionic degeneration, cortico-dentato-nigral degeneration, Rebeitz-Kolodny-Richardson syndrome), abnormalities involve both the basal ganglia and the cerebral cortex. Pathologically, there is asymmetric frontoparietal neuronal loss and gliosis, with swollen, achromatic neurons, tauimmunoreactive neuronal and glial inclusions, and nigral degeneration. Clinically, the corticobasal syndrome (CBS) is characteristically very asymmetric initially, with rigidity, bradykinesia, and occasionally tremor, accompanied by evidence of higher cortical dysfunction such as apraxia, agnosia, cortical sensory loss, focal myoclonus, or pyramidal signs. It has been increasingly recognized that not all patients with CBS have CBD pathologically. Many have other pathology, most commonly PSP or Alzheimer’s disease. Conversely, some patients with a clinical picture of PSP, frontotemporal dementia or Alzheimer’s disease may have CBD pathologically.
CBS typically begins with clumsiness, stiffness, or jerking of an arm. The alien limb phenomenon is common. There may be dystonic posturing as well as spontaneous and reflex myoclonus of the involved limbs. The involved limbs become stiff, jerky, and eventually useless. The combination of unilateral parkinsonism unresponsive to levodopa, accompanied by ideomotor apraxia of the involved extremities, is very suggestive. MRI frequently shows asymmetric cortical atrophy. Cognitive impairment may emerge as the disease progresses and becomes generalized. The disorder progresses slowly and does not respond to levodopa. Tau protein is a microtubule-associated protein, which when aggregated causes neurofibrillary tangles. Other conditions are associated with abnormalities of tau protein, especially PSP and frontotemporal dementia, collectively referred to as tauopathies.
Diffuse Lewy Body Disease
In diffuse Lewy body disease, the usual clinical picture is progressive dementia with added parkinsonian features in an elderly patient. It is the second most common degenerative dementia after Alzheimer’s disease. Parkinsonism may occur early or late and varies in severity. The parkinsonian features are typically more symmetric and milder than in PD. Tremor occurs but is less common and less severe than in PD. Another characteristic feature is psychotic behavior with visual hallucinations, delirium, and paranoia. Other common clinical features include cognitive fluctuations, dysautonomia, sleep disorders, especially REM sleep behavior disorder, and neuroleptic sensitivity. Functional imaging modalities such as resting state blood oxygen level-dependent functional connectivity MRI may assist in discriminating between diffuse Lewy body disease and Alzheimer’s disease.
Wilson’s Disease
Wilson’s disease (hepatolenticular degeneration, cerebral pseudosclerosis, Westphal-Strümpell syndrome) is a rare, autosomal recessive disorder due to abnormal copper deposition in the brain, especially the basal ganglia, liver, eye, and other tissues because of a genetic defect in an ATPase involved in copper transport (ATP7B), usually accompanied by a defect in the copper transport protein ceruloplasmin. The genetic defect leads to impaired copper excretion and systemic copper accumulation. A low ceruloplasmin level is found in the serum in 95% of patients. Ceruloplasmin is involved in the transfer of copper to copper-containing enzymes such as cytochrome oxidase, and the dysfunction of these enzymes may underlie the clinical manifestations.
The usual age of onset is between the ages of 10 and 20, and major manifestations include tremor, rigidity, dystonia, and abnormal involuntary movements of various types, dysarthria, dementia, parkinsonian features, spasticity, cerebellar signs, and psychiatric abnormalities (anxiety, depression, psychosis). The tremor may be present at rest and increased by voluntary movement. Most characteristic is a “wingbeating” tremor of the proximal upper extremities, consisting of a slow, high-amplitude, up-and-down movement of the elbow when the arm is held with the shoulder abducted and the elbow flexed. Pathologically, there is symmetric degeneration of the lenticular nuclei, with widespread neuronal loss and proliferation of Alzheimer type II astrocytes. Kayser-Fleischer rings are crescents of green-brown discoloration of the cornea due to copper deposits in Descemet’s membrane (Figure 30.3); these are essentially always present in patients with neurologic involvement but may not be visible without a slit lamp. Rarely, the disease may present without the rings. Slit lamp examination may also detect sunflower cataracts due to copper deposition in the lens. Risus sardonicus refers to an unusual forced “smile” due to facial dystonia and may be seen in Wilson’s disease and other disorders. Wilson’s disease may also cause excessive grinning, out of proportion to any amusement (for video, see Cetlin et al.). Other manifestations of the disease include cirrhosis, atypical hepatitis, hemolytic anemia, and renal disease. Patients may present with liver disease alone, brain disease alone, or evidence of both. In series of 282 patients seen over three decades, the mean age at diagnosis was 16, and the predominant features were neurologic (69%), hepatic (15%), hepatoneurologic (2.5%), osseomuscular (2%), pure psychiatric (2%), and asymptomatic (5%). The predominant neurologic symptoms were parkinsonism (62%), dystonia (35%), cerebellar (28%), and pyramidal signs (16%). Kayser-Fleischer rings were seen more commonly in those with neurologic symptoms compared with those with predominant hepatic presentations and those who were asymptomatic (100%, 86%, and 59%, respectively). Long-term treatment with penicillamine to chelate and remove the excess copper is beneficial (Figure 30.3). Copper deficiency myeloneuropathy may complicate long-term zinc therapy in Wilson’s disease. A similar myeloneuropathy may occur with zinc toxicity. An acquired form of hepatolenticular degeneration, unrelated to copper toxicity, may occur in patients with moderate to severe cirrhosis of the liver of various causes. A disorder with involuntary movements, dysarthria, and abnormal copper metabolism, not due to either Wilson’s or Menkes’ disease, may occur.
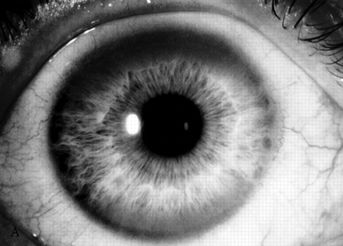
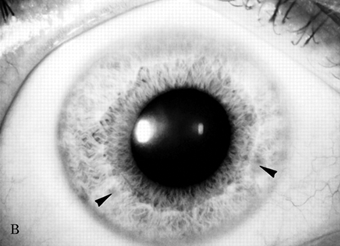
FIGURE 30.3 A. Marked Kayser-Fleischer corneal ring superiorly and inferiorly. B. Same eye 12 years after successful treatment with D-penicillamine. Note marked regression of Kayser-Fleischer corneal ring, which has virtually disappeared inferiorly. (Reprinted from Heckmann JG, Lang CJ, Neundorfer B, et al. Neuro/Images. Kayser-Fleischer corneal ring. Neurology 2000;54:1839, with permission.)
Pantothenate Kinase–Associated Neurodegeneration
Pantothenate kinase-associated neurodegeneration (PKAN, neurodegeneration with brain iron accumulation type-1, Hallervorden-Spatz syndrome) is a rare autosomal recessive disorder associated with macroscopic rust-brown discoloration of the globus pallidus and substantia nigra due to iron deposition. The clinical phenotype is variable. The disease usually begins in the first to the fourth decade of life, with rigidity, involuntary movements, ataxia, and dystonia, followed by pyramidal signs and progressive dementia. MRI findings are characteristic. T2-weighted sequences show bilaterally symmetric low signal intensity in the globus pallidus, due to iron deposition, surrounding a focus of high signal intensity, due to gliosis. This “eye of the tiger” image pattern is virtually diagnostic for PKAN.
Dentatorubropallidoluysian Atrophy
Dentatorubropallidoluysian atrophy (another Ramsay Hunt syndrome) is a heredofamilial degeneration, due to CAG repeats, in which the pathologic changes involve primarily the dentate, globus pallidus externa, red nucleus, and subthalamic nucleus. Clinical manifestations include choreoathetosis, dystonia, dementia, myoclonus, and ataxia; the disease is often included as one of the hereditary ataxia syndromes. Most reported cases have been from Japan. A cluster of cases occurred in the United States around the Haw River in North Carolina (Haw River syndrome).
HYPERKINETIC MOVEMENT DISORDERS
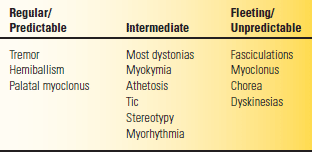
Hyperkinesia refers to increased movement. Hyperkinesias are abnormal involuntary movements that occur in a host of neurologic conditions. Hyperkinesias come in many forms, ranging from tremor to chorea to muscle fasciculations to myoclonic jerks. Any level of the motor system, from the motor cortex to the muscle itself, may be involved in their production. The only common characteristic is that the movements are spontaneous and, for the most part, not under volitional control. They may be rhythmic or random, fleeting or sustained, and predictable or unpredictable and may occur in isolation or accompanied by other neurologic signs. Table 30.2 summarizes some of these features.
TABLE 30.2 Abnormal Involuntary Movements as a Spectrum of Movements
In the examination of abnormal movements, the following should be noted: (a) the part of the body involved or the exact location of the movements; (b) the extent of the movement, or its distribution as it regards to part of a muscle, an entire muscle, movement involving joints, or more complex or composite patterns consisting of a sequence of different movements; (c) the pattern, rhythmicity, uniformity, multiformity, and regularity of recurrence—there may be a regular or rhythmic recurrence of activity involving the same muscle or groups, or there may be an irregular pattern of constantly changing motion of different parts; (d) the course, speed, and frequency of each particular movement; (e) the amplitude and force of the movement; (f) the relationship to posture, rest, voluntary activity or exertion, involuntary activity, various stimuli, fatigue, and time of day; (g) the response to heat and cold; (h) the relationship to emotional tension and excitement; (i) the degree that movements are suppressible by attention or the use of sensory tricks; and (j) the presence or absence of the movements during sleep. In general, involuntary movements are increased by stress and anxiety and decrease or disappear with sleep. Truly involuntary movements must be separated from complex or bizarre voluntary movements, such as mannerisms or compulsions.
It may be possible to name movements that fit a well-defined clinical pattern, but it is often better to describe the abnormality. Palpation may sometimes be useful, especially if the movements are very fine and limited to individual muscles. Videos are often very useful in the diagnosis and management of movement disorders.
TREMOR
A tremor is a series of involuntary, relatively rhythmic, purposeless, oscillatory movements. The excursion may be small or large and may involve one or more parts of the body. A simple tremor involves only a single muscle group; a compound tremor involves several muscle groups and may have several elements in combination, resulting in a series of complex movements (e.g., alternating flexion and extension together with alternate pronation and supination). Not only the agonist and antagonist, but muscles of fixation and synergists may play a part in the movements. A tremor may be present at rest or with activity. Some tremors are accentuated by having the patient hold the fingers extended and separated with the arms outstretched. Slow movements, writing, and drawing circles or spirals may bring tremor out.
Tremors may be classified in various ways: by location, rate, amplitude, rhythmicity, relationship to rest and movement, etiology, and underlying pathology. Other important factors may include the relationship to fatigue, emotion, self-consciousness, heat, cold, and the use of medications, alcohol, or street drugs. Tremor may be unilateral or bilateral and most commonly involves distal parts of the extremities —the fingers or hands—but may also affect the arms, feet, legs, tongue, eyelids, jaw, and head and may occasionally seem to involve the entire body. The rate may be slow, medium, or fast. Oscillations of 3 to 5 Hz are considered slow; 10 to 20 Hz, rapid. Amplitude may be fine, coarse, or medium. Tremor may be constant or intermittent, rhythmic or relatively nonrhythmic, although a certain amount of rhythmicity is implied in the term tremor. Irregular “tremor” may be due to myoclonus.
The relationship to rest or activity is the basis for classification into two primary tremor types: rest and action. Resting (static) tremors are present mainly during relaxation (e.g., with the hands in the lap), and attenuate when the part is used. Rest tremor is seen primarily in PD and other parkinsonian syndromes. Action tremors appear when performing some activity. Action tremors are divided into subtypes: postural, kinetic, task-specific, and isometric. Only when they are very severe are action tremors present at rest. Postural tremors become evident when the limbs are maintained in an antigravity position (e.g., arms outstretched). Common types of postural tremor are enhanced physiologic tremor and ET. Kinetic tremor appears when making a voluntary movement, and may occur at the beginning, during or at the end of the movement. The most common example is an intention (terminal) tremor. Intention tremor is a form of action tremor seen primarily in cerebellar disease (see Chapter 43). The tremor appears when precision is required to touch a target, as in the finger-nose-finger or toe-to-finger test. It progressively worsens during the movement. Approaching the target causes the limb to shake, usually side-to-side perpendicular to the line of travel, and the amplitude of the oscillation increases toward the end of the movement. Some tremors fall into more than one potential classification. Most tremors are accentuated by emotional excitement, and many normal individuals develop tremor with anxiety, apprehension, and fatigue. A shivering type of tremor (rigors) may be brought on by cold, but identical movements can be psychogenic.
Physiologic tremor is present in normal individuals. The frequency varies from 8 to 12 Hz, averaging about 10 Hz in the young adult, somewhat slower in children and older persons. The frequency for an individual is the same at different sites in the body. The visible tremor brought out in normal persons by anxiety, fright, fatigue (rock climber’s tremor, Elvis leg), and other conditions with increased adrenergic activity is accentuated or enhanced physiologic tremor. A typical example of enhanced physiologic tremor is that seen in hyperthyroidism. The tremor involves principally the fingers and hands, and may be fine and difficult to see. It may be brought out by placing a limb in a position of postural tension, by performing voluntary movements at the slowest possible rate, or holding the index fingertips as close together as possible without touching. The tremor may be better appreciated by placing a sheet of paper on the outstretched fingers; shaking of the paper may be obvious even though tremor is not grossly visible. Physiologic tremor may be present both at rest and on activity, but it is accentuated by activity as well as by anxiety and emotional stress. Similar tremor occurs due to the effects of alcohol, nicotine, caffeine, amphetamines, ephedrine, and other stimulants (Table 30.3). A fine tremor of the closed eyelids is seen in hyperthyroidism (Rosenbach’s sign).
TABLE 30.3 Some Drugs that Cause Tremor





