TYPICAL ABSENCE SEIZURES
Clinical Features
Typical absence seizures last an average of 10 seconds, are usually provoked by hyperventilation, and postictal confusion is either absent or very brief (7,8). The International League Against Epilepsy (ILAE) classification recognizes seven subforms of typical absences (2):
- Absence with impaired consciousness only
- Absence with mild clonic components
- Absence with atonic components
- Absence with tonic components
- Absence with automatisms
- Absence with autonomic phenomenon
Most patients exhibit a mix of these subforms. In the majority of absence seizures, the first clinical sign is arrest of activity, but other initial signs include eyelid movements, eye opening, altered awareness, or automatisms. As the seizure evolves, other clinical signs may develop. In a clinical and EEG study including 339 typical absence seizures in 47 children, the first clinical sign occurred at an average of 1 second into the seizure (SD 1.29, range 0 to 9 seconds), and included arrest of activity (46%), eyelid movement (28%), eye opening (19%), altered awareness (7%), or automatisms (4%) (9). Most children who were engaged in an activity prior to the episode had an arrest of activity, though 8% continued their activity. If awake with eyes initially closed, the eyes opened in 59% of all the seizures and in at least one seizure in 95% of children. Some children opened their eyes at the beginning of the seizure and others a few seconds into the seizure. Staring was seen in 78% of children who had their eyes open. The direction of the eye stare tended to be consistent in an individual child, but varied among children. Consistent myoclonic movements involving muscles of the head, face, trunk, and limbs were seen in four children. Automatisms were seen in 41% of seizures and were mostly oral (72%). The child was unaware of the automatisms in 75% of cases and partially aware in 20%. Response testing revealed that 75% were completely unaware, with no response to testing and no memory of the events, during at least part of the seizure, and an additional 20% were partially unaware.
In untreated patients, absence seizures are frequent and are quickly documented on routine EEG testing, with most seizures occurring within 10 minutes of EEG recording, and more than 90% of seizures occurring before or during a trial of hyperventilation (8).
TYPICAL ABSENCE SEIZURES
EEG Features
Absence seizures are characterized electrographically by bilateral, synchronous, symmetric spike-and-wave discharges at a frequency of approximately 3 Hz, a finding first published in 1935 by Frederic Gibbs and William Lennox (10) (Fig. 15.1).
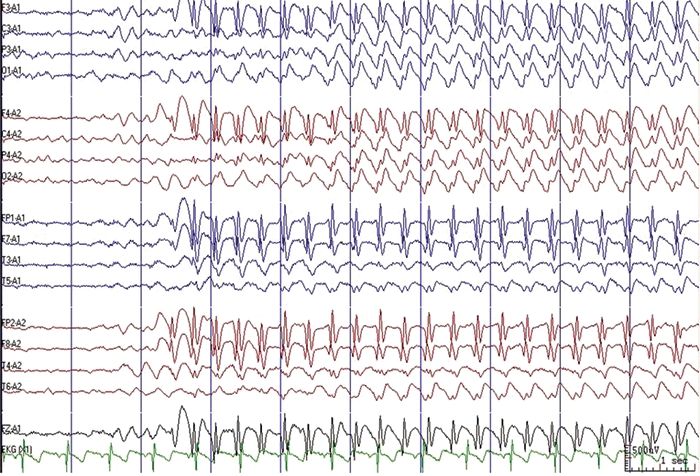
Figure 15.1. EEG during hyperventilation showing generalized, 3 Hz spike-and-wave discharge. Use of an ipsilateral ear reference montage often provides a clear display of spike-and-wave morphology.
In a recent randomized clinical trial of newly diagnosed patients with CAE (5,8,11), the median time to first seizure across 440 patients on a pre-treatment EEG was 6.0 minutes (range 0 to 59 minutes). The median number of seizures per 1-hour study EEG was 5 (range 1 to 60), and the median seizure duration was 10.8 seconds (range 3.3 to 77.6 seconds). The median duration of shortest seizure per EEG was 7.5 seconds (range 3 to 77.6 seconds), whereas the median longest seizure per EEG was 14.9 seconds (range 3.6 to 105.3 seconds). At least one seizure lasting 20 seconds or longer was noted in 29% of subjects (129/440).
Generalized spike wave (GSW) frequency at seizure onset is typically 3 Hz, with a range of 2.7 to 4.0 Hz. Toward the end of the discharge, the frequency often slows and the spikes become less obvious or disappear. The electrical field of the GSW burst is maximally negative at the bifrontal or frontopolar electrodes. Within a sample of 721 GSW bursts reviewed by three EEG readers for morphologic characteristics, 84% (604/721) of bursts contained single spike-and-wave discharges at onset, 3% (21/721) contained polyspike-and-wave discharges at onset, and consensus on spike morphology could not be reached with 13% of bursts (96/721) (8). Since only 3% of bursts contained definite polyspikes and 13% of bursts contained unclear spike morphology, usually because of inconsistent morphology based on electrode location or montage, GSW morphology may not be a reliable and reproducible variable to consider when evaluating EEG features and treatment response, later evolution of other seizure types, or outcome.
EEG variations may be present in typical absence seizures. The ictal onset can be asymmetric for about 0.5 seconds prior to generalization on scalp EEG. While most bursts are frontally predominant at onset, some are predominant in the occipital regions. Unilateral discharges are very rare in untreated patients, but can be seen occasionally once treatment is begun. Fragments of GSW bursts are also seen, and are especially common after treatment is begun. Photoparoxysmal response is uncommon in typical absence seizures, seen in only 1% to 15% of patients (8,12).
The interictal EEG in most children with CAE is usually normal, unless short fragments of GSW bursts are present. Rhythmic posterior bilateral delta activity, ranging from 2.5 to 4 Hz, is seen in 21% to 32% of patients (8,9); frontal intermittent rhythmic delta activity in 4.5%; focal sharp waves (distinct from fragments of GSW) in 2.5%; and focal slowing in 0.7% (8). During sleep, GSW bursts become more irregular in frequency and morphology, with polyspike and wave discharges seen in 40% of children (9).
ATYPICAL ABSENCE SEIZURES
Clinical Features
Patients with atypical absence seizures often have a structural or metabolic (remote symptomatic) etiology for their epilepsy, and baseline rates of developmental delay and intellectual disability are as high as 95% (6). Since typical and atypical absence seizures can both include complex automatisms, incontinence, and changes in tone (13), a classification of atypical absence seizures should not be based on seizure semiology alone, but rather on a combination of patient factors, ictal semiology, and EEG features. In general, atypical absence seizures have a more insidious onset and offset, are longer in duration, include hypertonic or atonic components, and are less likely provoked by hyperventilation than typical absence seizures (2,6).
EEG Features
Typical and atypical absences share many similar clinical and EEG features, and should be considered as a part of the same continuum (14). Patients with atypical absence seizures have a more heterogeneous ictal recording, often with unclear ictal onset and offset, asymmetric spike-wave discharges, GSW frequencies of 2.5 Hz or less, and are less likely to activate with hyperventilation (2,6). Ictal duration can be unusually brief (<3 seconds) or long (>60 seconds). Interictal abnormalities often include background slowing and disorganization, focal or multi-focal slowing, focal or multi-focal sharp waves, and fragmentary and asymmetric spike-and-slow-wave complexes (2,6).
EPILEPSY SYNDROMES
Typical absence seizures are a core feature of at least five epilepsy syndromes: CAE, JAE, JME, MAE, and LGS.
CAE is defined by frequent daily absence seizures in normal school age children with EEG showing bilateral, synchronous, symmetric, approximate 3-Hz spike wave discharges, superimposed on a normal EEG background (2,5).
JAE occurs in older children, with onset between ages 8 and 16 with a peak age between 10 and 12 years (15,16). The syndrome includes absence, generalized tonic clonic seizures (GTCs) and myoclonic seizures and typically requires long-term treatment. The absences in JAE can be frequent, although usually less frequent than CAE, and are typically not triggered by hyperventilation (2,5,17).
In JME, absence seizures occur in about one-third of patients and are usually infrequent and not associated with automatisms, making an accurate absence seizure count challenging to obtain. They generally respond well to treatment, although long-term treatment is usually required (2,17). The overlap between JAE, JME, and primary GTCs upon awakening can be substantial, but the presence of myoclonic seizures should prompt careful consideration of a diagnosis of JME.
Atypical absence seizures are seen in MAE and LGS. Both syndromes occur in children with associated neurodevelopmental disabilities and other seizure types. Absences in MAE are often associated with rhythmic myoclonic jerks, and are treatment-resistant (2,17). In LGS, tonic and atonic seizures, plus atypical absence seizures, are the core seizure types. Absence seizures in LGS are challenging to monitor, since clinical signs can be subtle and the children typically have coexistent cognitive and behavioral disabilities.
Eyelid myoclonia with absences (Jeavons syndrome) includes typical absence seizures with the usual ictal signature of approximate 3 Hz GSW and episodes of rapid eye flutter with retention of awareness (eyelid myoclonia). In Jeavons syndrome, the eyelid myoclonia typically correlates with GSW or polyspike and wave discharges on EEG. Some patients with absence seizures have paroxysmal eyelid movements with no ictal change on scalp EEG other than eye flutter artifact. This condition can be easily confused with Jeavons syndrome, and differentiation between the two requires EEG. However, neither the ictal eyelid myoclonia of Jeavons syndrome nor the paroxysmal eyelid movements sometimes associated with absence seizures typically respond to treatment with antiepileptic drugs (18–20).
Absence status epilepticus, which can involve typical or atypical absence seizures, is defined as prolonged absence seizures lasting more than half an hour, with continuous impairment of consciousness and generalized discharges of 1 to 4 Hz spikes or polyspikes and slow waves on the EEG. Absence status can be a seemingly unprovoked exacerbation of an underlying seizure disorder or may be associated with inappropriate antiepileptic drug choice, drug intoxication, drug withdrawal, or electrolyte disturbance (21). Once absence status is successfully treated or stops spontaneously, the patient typically returns to baseline without deleterious effects, as long as injuries are avoided.
Pathogenesis
Absence seizures are provoked by an abnormal thalamo-cortical circuitry that activates abnormal oscillatory rhythms, which then generate 3 Hz spike-and-wave discharges (22). The cellular mechanism involves low-current T-type calcium channels, which are blocked by ethosuximide (ESM) (17). GABAB receptors appears to play a role in the generation of absence seizures as suggested by the observation that GABAB receptor agonists worsen and GABAB receptor antagonists suppress absences (22).
Genetic factors play an important role in the pathogenesis of typical absence seizures, as suggested by a high concordance rate among twins (23). Mutations in genes that encode GABAA receptor (24), voltage-gated chloride channels (25), and T-type calcium channels (26) have been identified. However, the vast majority of patients with absence seizures do not have an identifiable genetic cause at this time.
About 10% of patients with early-onset absence epilepsy, defined as onset age before 4 years of age, have glucose transporter 1 (GLUT1) deficiency caused by a mutation in SLC2A1. GLUT1 is a protein that facilitates the transport of glucose across the blood–brain barrier (27,28). GLUT1 deficiency can be diagnosed by gene testing or by the presence of hypoglycorrhachia (fasting CSF glucose <2.2, CSF/plasma glucose ratio <0.45) (27). This diagnosis is important because seizures caused by GLUT1 deficiency can be treated with the ketogenetic diet. SCL2A1 mutations do not appear to play a major role in absence epilepsies beginning after the age of four (28).
Differential Diagnosis
Absence seizures may be confused with normal daydreaming, inattentiveness associated with ADHD, tics with eye blinking or facial movements, or focal seizures with impairment of awareness. However, in a child with absence seizures, a careful history will usually reveal stereotypic episodes of arrest of activity, decreased responsiveness, associated ocular, facial or extremity automatisms, and a quick return to baseline. Isolated staring episodes without other features are almost never absence seizures, but rather reflect normal behavior or inattentive ADHD. Clinical signs suggesting that the staring episodes are nonepileptic include: the events do not interrupt play; the events were first noticed by a professional such as a teacher, rather than a parent; and the staring child is responsive to touch or “interruptible” by other external stimuli (Table 15.2). Each of these features has close to 80% specificity for suggesting nonepileptic staring episodes. Twitches of the arms or legs, incontinence, or upward eye movement are more likely to be seen in epileptic seizures (29).
Table 15.2 Clinical Signs Suggesting that Staring Episodes are Nonepileptic. Specificity is the Percentage of Children without Epilepsy who are Correctly Identified as Not Having Epilepsy

Diagnostic Evaluation
For most children, routine EEG will confirm the diagnosis of untreated absence seizures within 10 minutes, if a trial of hyperventilation is performed early in the EEG recording (8). In the CAE treatment trial, only 6% of patients required more than 30 minutes of EEG recording to confirm the diagnosis, and 92% of patients had seizures before or during the first trial of hyperventilation (8). Once treatment has been initiated, treatment response should be assessed by clinical history, office-based trials of hyperventilation, and routine EEG, since approximately 30% of patients who are seizure-free by parent-teacher report have seizures on a 1-hour EEG recording (12).
Treatment
Based on the results of the CAE treatment trial, published in 2010, ESM has class I evidence as the optimal initial treatment for CAE (5). This double-blind, randomized, controlled trial enrolled 453 children with newly diagnosed and untreated CAE, who were randomly assigned to treatment with ethosuximide (ESM, n = 156), lamotrigine (LTG, n = 149), or valproic acid (VPA, n = 148). The primary outcome measure was freedom-from-failure (FFF), defined as no clinical or electrographic seizures at 16 to 20 weeks of therapy and no treatment-limiting adverse effects. The FFF rates for ESM and VPA were similar (53% and 58%, respectively, P = 0.35) and were higher than the FFF rate for LTG (29%, P < 0.001 when compared to both ESM and VPA). Discontinuation rates due to adverse events were not significantly different between the three drugs. However, post-treatment attentional dysfunction was more common with VPA than with ESM (49% and 33%, respectively, P = 0.03). The study concluded that while ESM and VPA were more effective than LTG in the treatment of CAE, ESM is the optimal initial treatment for CAE as it was associated with fewer adverse attentional effects.
At 12 months of follow-up, ESM and VPA remained superior to LTG, but the VPA group experienced higher rates of drug discontinuation and attentional dysfunction. VPA-associated weight gain also became apparent at 12 months follow-up (12). Across all three treatment groups, only 37% of all subjects continued on their first medication at 12 months. FFF rates for ESM and VPA were similar (45% and 44%, respectively, P = 0.82) and were higher than LTG (21%, P < 0.001 when compared to both ESM and VPA). Almost two-thirds of the 125 subjects with treatment failure due to lack of seizure control were in the LTG group. The largest subgroup (42%) of the 115 subjects discontinuing due to adverse events was in the VPA group. The previously reported higher rate of attentional dysfunction seen at 16 to 20 weeks in the VPA group compared with the ESM or LTG groups persisted at 12 months (P < 0.01).
Strikingly, even the best initial monotherapy fails in 55% of children with CAE over the first 12 months (12). Overall, ESM provides the best, though still very suboptimal, combination of efficacy and tolerability, and does not worsen inattention. VPA is as effective as ESM for seizure control, but has higher rates of adverse effects and worsens inattention. LTG is not as effective in controlling seizures, but is well-tolerated and does not worsen inattention. If ESM fails, then either VPA or LTG are possible second-line choices depending on seizure burden, attentional difficulties, body mass index, and other factors. If all three drugs fail in monotherapy, then combination therapy can be considered (17), although high-quality evidence to support its use is lacking.
Other medication treatment options for CAE, either alone or in combination, based on case series and case reports, include zonisamide (30); benzodiazepines, particularly clonazepam (16); acetazolamide (13); levetiracetam (31); or felbamate (32). Ketogenic diet (33), vagus nerve stimulation (34), and amantadine (35) have also been reported in small case series to have efficacy in some patients with treatment-resistant absence seizures.
Importantly, carbamazepine, vigabatrin, and tiagabine are contraindicted in the treatment of absence seizures, since these agents have been reported to worsen absence seizures and precipitate absence status epilepticus (7,21,36).
Prognosis
Seizure Outcomes
In the CAE treatment trial, longer pre-treatment seizure duration was associated with better drug response outcome at 16 to 20 weeks. Children whose shortest seizure lasted longer than 7.5 seconds had better response to initial treatment than those with briefer seizures. However, patients with any seizures longer than 20 seconds were more likely to display attentional difficulties (8). These findings underscore the complex relationship between seizures and inattention in CAE.
Reported long-term remission rates for absence seizures in CAE range from 56% to 84% (33,37). In small series, factors associated with persistent seizures past puberty include onset after 8 years of age, GTCs at the time of absence seizures, myoclonic jerks, eyelid or perioral myoclonia, family history of GTCs, absence status, and background slowing on EEG (38,39). Focal interictal spikes have not been shown to be related to outcome (40).
A retrospective cohort study of 115 patients found that age of onset >8 years and family history of GTCs in patients with CAE predict development of GTCs, but polyspike and wave discharges do not. It is possible that CAE with GTCs is a distinctive syndrome from CAE without GTCs (41).
One retrospective chart review of 119 patients found that typical absence seizures are more likely to respond to the initial antiepileptic drug compared to atypical absences, but the remission at 2 years is similar in the two groups (14).
There are few outcome studies of JAE. GTCs are seen in 47% to 95% of patients, and remission rates range from 37% to 62% (15,39,42,43). Some studies (15,42) have found that myoclonus and GTCs are associated with persistent seizures, but other studies have not confirmed this finding (39). Another case series of CAE and JAE found that patients who responded to initial AED therapy were more likely to achieve remission and not progress to JME (38).
Psychosocial Outcome
CAE is often described as a benign epilepsy, with few associated cognitive and behavioral challenges. However, there is a growing evidence suggesting that the psychosocial impact of CAE is grossly underestimated and the need for early detection and treatment of comorbid cognitive and psychological difficulties (37,44,45). In the CAE treatment trial, baseline rates of inattention were 30% to 42%, inattention did not improve even with successful seizure treatment, and VPA worsened inattention. This finding dismisses the notion that successful treatment of seizures also improves comorbid inattention in CAE.
Children with CAE, when compared to matched controls, also have increased risk of cognitive deficits (25% of CAE children), linguistic deficits (43%), psychiatric diagnoses (61%), particularly ADHD and anxiety disorders, and clinically relevant scores (30%) on a child behavior checklist. Only 23% of the patients in this study had interventions directed at these co-morbid conditions (44).
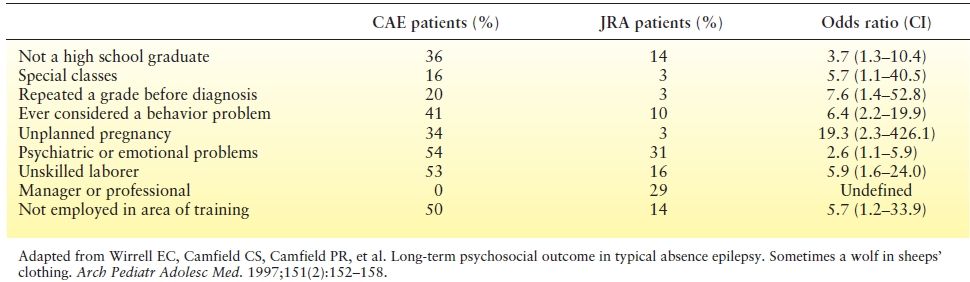
Long-term outcome is also very concerning. A population- based study from Nova Scotia found that young adults with a history of typical absence seizures had greater psychosocial difficulties later in life compared to children with a non-CNS chronic disease (juvenile rheumatoid arthritis). Findings are summarized in Table 15.3. On every measure, young adults with a history in CAE fared worse than young adults with JRA (37). Most patients in this study had entered remission of their seizures and, while persistence of seizures predicted even worse psychosocial outcomes, remission of seizures did not ensure favorable psychosocial outcome (37).
Table 15.3 Psychosocial Outcome of Patients with CAE Compared with JRA in their Mid-20s
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


