72 Acquired Polyneuropathies
Diagnostic Approach
Clinical Vignette
The evaluation for the etiology of a patient’s polyneuropathy can be very challenging for many reasons, including the fact that there are more than 100 potential etiologies (Fig. 72-1). Ultimately the polyneuropathy is determined to be acquired (i.e., caused by some other disease or exposure) in one third of cases (Box 72-1), inherited in another one third of cases (see Chapter 71), and—in spite of appropriate testing—idiopathic in the remaining one third of cases. In order to focus on a smaller list of potential etiologies so that the evaluation can be simplified, we believe that it is best for the clinician to first characterize the polyneuropathy and the patient. We will present one method for characterizing neuropathy that is easy to remember, based on four simple clinical questions about the neuropathy and the patient: “What?” “Where?” “When?” and “What setting?”
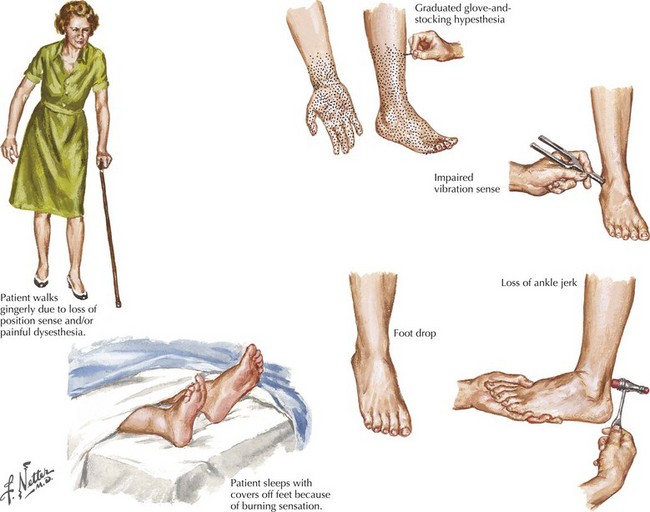
“What?” refers to what nerve fiber modalities (motor, sensory, autonomic, or a combination) are involved? The identification of sensory nerve involvement, at a minimum, allows the clinician to exclude other neuromuscular diseases not associated with sensory dysfunction, such as disorders of anterior horn cells (e.g., amyotrophic lateral sclerosis), neuromuscular transmission (e.g., myasthenia gravis), or of muscle (myopathy). When sensory symptoms and signs are present, it is useful to characterize neuropathic sensory symptoms into “positive” or “negative” because acquired neuropathies are usually accompanied by positive neuropathic sensory symptoms and inherited neuropathies are usually not. Positive sensory symptoms may be painful (e.g., “burning,” “freezing,” or “throbbing”) or painless (e.g., “tingling” or “swelling”) (Box 72-2). Paresthesias and pain (positive neuropathic sensory symptoms) are common complaints for patients with diabetic, vasculitic, alcoholic, or uremic neuropathy and patients with Guillain–Barré syndrome (GBS) or chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). In the clinical vignette presented above, the patient had prominent positive neuropathic sensory symptoms, strongly suggesting that the etiology of her neuropathy is acquired rather than inherited. Patients with neuropathy often also complain of exaggerated discomfort to painful sensory stimuli (hyperalgesia) and to nonpainful sensory stimuli (allodynia). Patient complaints indicative of negative neuropathic sensory symptoms include loss of sensation and imbalance (i.e., sensory ataxia). Most patients with neuropathy have some degree of motor nerve involvement that at times is overshadowed by the sensory complaints. Our patient had prominent motor nerve fiber involvement.
Box 72-2 Painful Polyneuropathies*
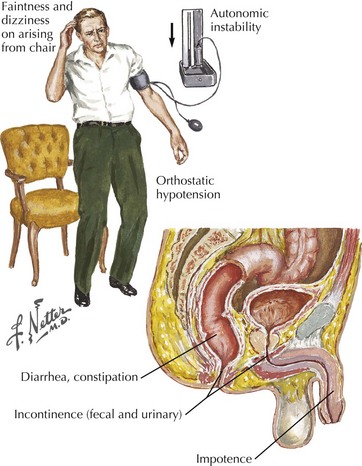
Identification of autonomic nerve involvement can be an important clue because only a small number of neuropathic processes affect both autonomic and somatic nerves (e.g., GBS, paraneoplastic neuropathy, diabetic neuropathy, amyloid neuropathy) (Box 72-3). Autonomic symptoms include lightheadedness, syncope, diarrhea, constipation, postprandial bloating, early satiety, urinary complaints, erectile dysfunction, abnormal or absent sweating, and dry mouth and eyes (Fig. 72-2). Our patient did not have discernible autonomic nervous system involvement.
Box 72-3 Polyneuropathies with Autonomic Nervous System Involvement
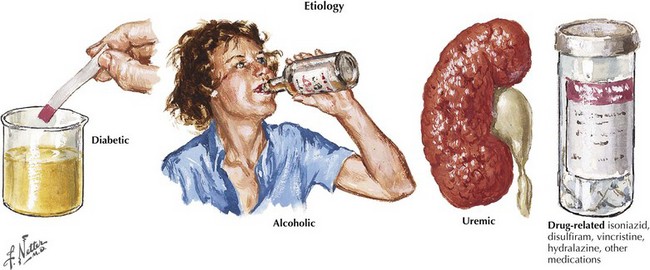
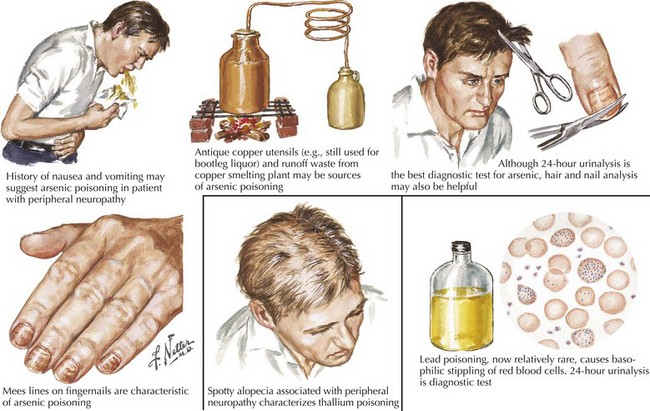
“What setting?” refers to an elaboration of the unique clinical circumstance of the individual patient. This is done by determining what in the patient’s past medical history, medication list, social history, family history, and the review of systems may be relevant (Fig. 72-3). An understanding of the significance of these clinical factors requires knowledge of the risk factors of neuropathy and knowledge of the clinical features of the diseases that may be risk factors for neuropathy. For example, unexplained weight loss raises concern for vasculitis or malignancy (e.g., small cell lung cancer), both of which cause an immune-mediated neuropathy. The neuropathy secondary to malignancy (e.g., paraneoplastic neuropathy) usually presents differently than vasculitic neuropathy, so it is usually not too difficult to differentiate these two etiologies. A clinical setting of known diabetes mellitus or known kidney disease would elevate those comorbidities on the differential diagnosis. Heavy metal poisoning or other intoxication, although rare, needs to be considered in the patient with systemic symptoms (e.g., nausea, vomiting) and other manifestations suspicious for poisoning (Fig. 72-4). Our patient’s presentation is of vasculitic neuropathy rather than paraneoplastic neuropathy.
The fifth step in characterization requires NCS and EMG. NCS and EMG can contribute to (or rarely refute) the clinical characterization in terms of “what” and “where,” as well as provide another view of the temporal evolution (“when”). NCS and EMG can also characterize the neuropathy as being primarily axonal or demyelinating. Neuropathies with axonal injury are far more common than those primarily with demyelination, but the identification of a primarily demyelinating polyneuropathy is very important because acquired demyelinating polyneuropathies (e.g., GBS, chronic inflammatory demyelinating polyradiculoneuropathy, multifocal motor neuropathy) are generally treatable. They are usually immune-mediated and treatable with immunotherapy (e.g., corticosteroids, intravenous immunoglobulin [IVIG], plasmapheresis) (Box 72-4). NCS and EMG can also assess for subclinical involvement and provide baseline parameters in case future testing is necessary to monitor the patient’s course. Our patient’s NCS and EMG confirmed multiple, axonal mononeuropathies. It is important to note that the localization of each mononeuropathy was not at common sites of nerve entrapment, such as the elbow for the ulnar nerve and the fibular head for the peroneal nerve; thus, a compression or entrapment mechanism of injury is not plausible (given the other facts of the case, it would have been very unlikely anyway).
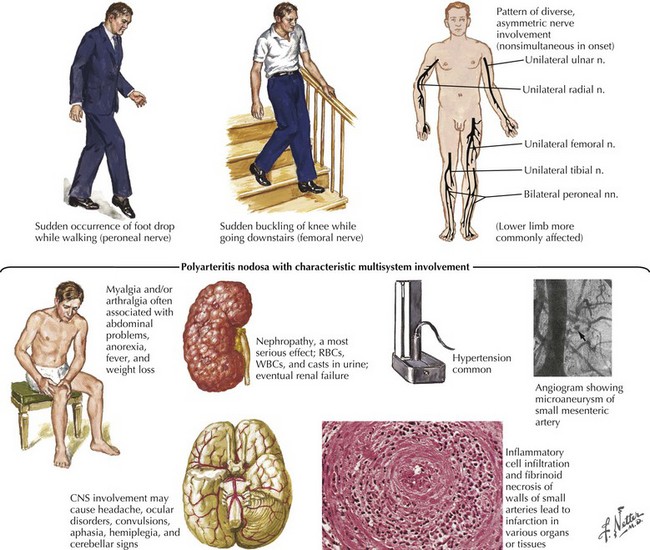
Vasculitis is only one of the more than 100 different causes of neuropathy (Fig. 72-5). It is a far less common cause of acquired neuropathy than diabetic, alcoholic, or uremic neuropathy, but it is very important to diagnose—and to do so quickly—because undiagnosed and untreated systemic vasculitis may be fatal. See discussion on vasculitic neuropathies below.
Idiopathic Length-Dependent Polyneuropathies
Polyneuropathies are one of the most common neurologic disorders; the length-dependent pattern is the most prevalent. Although there are many recognized causes of polyneuropathy, specific mechanisms have not been identified in 30–40% of patients, and these patients are deemed to have idiopathic neuropathy (Box 72-5).
Box 72-5 Length-Dependent Polyneuropathies
Dysimmune/Inflammatory
Pure sensory neuropathies fit into the LDPN pattern such as with diabetes mellitus, renal disease, vitamin deficiencies, various toxins, Sjögren syndrome, and amyloidosis (Fig. 72-6






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


