Figure 75.1. (A). Sagittal T1 sequence showing increased spinal cord diameter at C6-C7 and hypointense images within the cord suggesting the presence of vascular structures. (B). Sagittal T2 sequence of the same patient showing abnormal vascular structures within the cervical spinal cord (intramedullary vascular malformation).
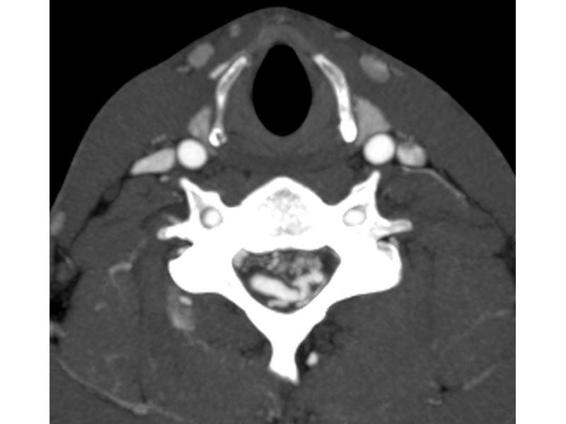
Figure 75.2. CT-angiography (CTA) of the same patient as in Figure 75.1. At the cervical level, abnormal and dilated vascular structures are seen intra- and extramedullary, corresponding to a cervical arteriovenous malformation.
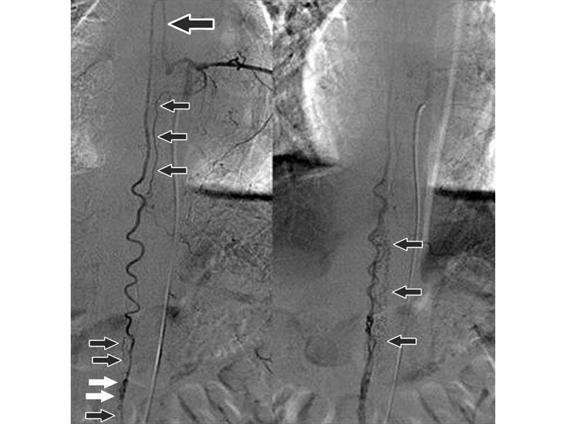
Figure 75.4. Digital angiography in a patient with an arteriovenous malformation (arrows) within the spinal canal at the levels of the chest and abdomen.
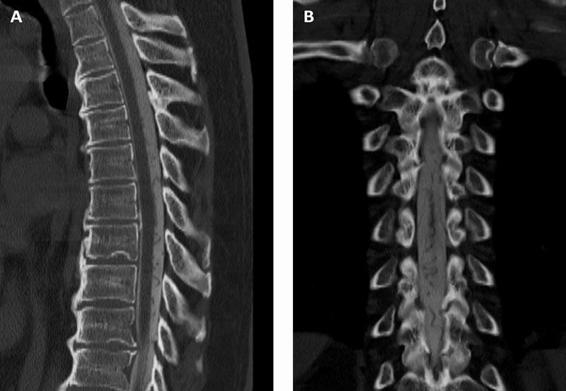
Figure 75.3. Tomographic myelogram showing serpiginous vascular structures in the extramedullary space corresponding to a dural arteriovenous fistula. (A). Sagittal reconstruction. (B). Coronal reconstruction.
75.2.4 Acute Transverse Myelitis
Acute transverse myelitis (ATM) is a syndrome of multiple neurologic deficits due to a focal inflammatory attack of the spinal cord. Although the name may imply the pathologic process was not present until symptoms arose, ATM is often the clinical presentation of a pathologic process that was previously unrecognized, subacute, acute or chronic or secondary to another disease. Although ATM can have vascular and structural aetiologies, our focus will be on pathologic processes where inflammatory activity leads to focal demyelination or destruction of the spinal cord. Publications often state that ATM has a bimodal distribution during the first and third decades of life. In reality, this is not absolute. As we improve in diagnosing patients with multiple sclerosis, Devic’s disease (neuromyelitis optica) and autoimmune-associated transverse myelitis, we encounter patients who can develop spinal cord lesions at any point during their disease. As an acute attack evolves and maximal deficits are reached, up to 50% of patients will develop motor paralysis and 80-95% will experience sensory disturbances below the level of the lesion. The diagnosis of ATM is often secondary to another disease or medical history:
- Multiple sclerosis.
- Devic’s disease (neuromyelitis optica).
- Recent infection.
- History of radiation exposure.
- Post-vaccination.
- Autoimmune-associated systemic illness.
- Neoplastic (paraneoplastic disorder or neoplasm causing compressive myelopathy).
- Idiopathic.
Since the clinical presentation will depend on multiple factors, no pathognomic sign or syndrome exists. A patient may present with his or her own distinct constellation of symptoms. When gathering the initial history of symptom presentation, “the tempo” will help differentiate inflammatory from vascular aetiology. Maximal clinical deficits should rapidly occur with vascular lesions, whereas inflammatory lesions can take up to 7 days to peak. Acute transverse myelitis occurs mostly in the thoracic region but cervical and lumbar segments are also vulnerable. Inflammatory lesions are not confined by anatomical or vascular boundaries; therefore, patients can present with a mixture of deficits in motor, sensory and autonomic function due to the compact anatomical arrangement of the spinal cord. Inflammatory ATM is often the end result of another disease process or seen for the first time in a “previously healthy” patient; hence, the past medical history, review of systems, and family history should be explored in detail. The review of system should be directed towards episodes of transient weakness, sensory disturbance, episodes of visual changes, recent febrile illness or contact with an ill person, Uhthoff’s sign (worsening of neurologic symptoms during exercise or excess body heat), dry mouth or dry eyes. The family history is important to look for associated demyelinating or autoimmune diseases that run in the family. Another important feature on exam is Lhermitte’s sign (electrical sensation running down the spine with flexion of the neck). On clinical exam, most patients will have a sensory level and sensory disturbance below the level of the lesion. In addition to motor weakness and sensory disturbance, autonomic symptoms such as bowel or bladder incontinence, urinary urgency, inability to completely void the bladder and sexual dysfunction can be present.
Once a complete neurologic exam has been performed and a patient demonstrates myelopathic features, an MRI with contrast should be one of the first actions taken. If MRI is contraindicated, then a CT-myelogram of the spinal cord can help to rule out compressive lesions. The first question to answer with neuroimaging is to rule out a compressive structural or vascular lesion that correlates with clinical symptoms on exam. If MRI demonstrates a compressive or vascular lesion, a neurosurgical consultation should be obtained immediately. In ATM, sagittal T2-weighted sequences demonstrate focal area(s) of hyperintensity often associated with an increase in the calibre of the spinal cord due to inflammation and edema. If focal, isointense areas of atrophy are noted; this may suggest previous episodes of transverse myelitis. To fully evaluate a focal hyperintensity, both the axial and sagittal T2 image sequences should be scrutinized simultaneously. Inflammatory lesions usually affect both the gray and white matter of the cord, involving up to half the diameter of the cord, and are often eccentric. Hyperintense lesions specific to multiple sclerosis should only span roughly two vertebral bodies. Lesions spanning more than three vertebral bodies imply either systemic disease associated ATM or Devic’s disease. In addition to neuroimaging of the spine, MRI with contrast of the brain is also useful to understand if the current acute illness is part of a larger process. Inflammatory lesions are a dynamic process; therefore, if the first MRI obtained is “non-diagnostic”, a repeat MRI 7 days later should be performed (Figure 75.5). Finally, if both brain and spine MRI only demonstrate spinal cord lesions, visual evoked potential (VEP) is still valuable to detect occult central nervous system (CNS) demyelination.
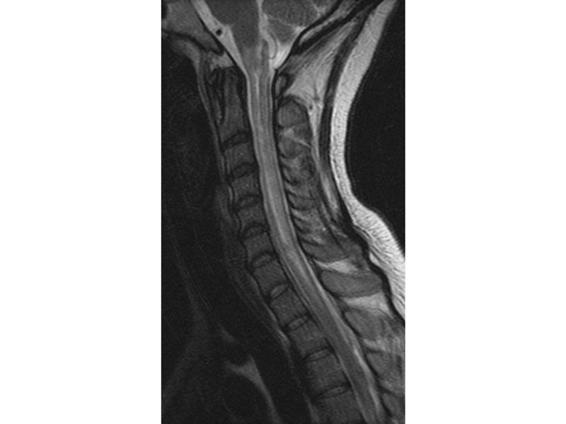
Figure 75.5. MRI, sagittal T2 sequence. Patient with ATM: extensive hyperintense area (predominantly central) involving the medulla oblongata, the whole cervical spinal cord, and the first segment of the thoracic cord.
Diagnosis and management of patients with ATM begin with basic serology, specialized serology and CSF analysis as listed below. Basic serology is meant to establish a baseline prior to immune-directed therapy and to rule out infection. In fact, in patients with a known history of demyelinating disease, an infection could have been the trigger for the ATM attack. Specialized serology is important for two groups of patients: first, for patients with known systemic autoimmune disease and, second, for patients with no significant past medical history. For example, in patients with known systemic lupus erythematosus (SLE) it is important to establish if their disease is active or dormant in relation to the attack. The second and most important group of patients are those considered “previously healthy” yet harbouring an occult disease. Swift pursuit of the underlying aetiology has both immediate and long-term implications when treating ATM. For example, in a small group of patients with Sjögren’s syndrome-associated ATM, their SS-A and SS-B serum titers can be negative on initial presentation. These patients require salivary gland lip biopsy to establish the diagnosis; however, if the biopsy is taken after multiple days of corticosteroid therapy, the biopsy results may return non-diagnostic. Furthermore, if the ATM occurred in a setting of occult autoimmune disease, then long-term treatment should incorporate management of the new diagnosis. Basic CSF studies in patients with ATM should demonstrate high protein levels and very mild pleocytosis due to breakdown of the blood-brain barrier. The remaining panel of viral PCRs (polymerase chain reaction) should be considered as well. Tests that should be performed are summarized in Table 75.2.
Basic serology |
|
Specialized serology |
|
CSF analysis |
|
Table 75.2. Serological tests.
All of these studies are aimed to help the clinician tailor a treatment regimen that will ultimately involve immunosuppression. Historically, when treating ATM, corticosteroid therapy has been the mainstay and should begin immediately once concurrent infection has been ruled out. The CBC, urinalysis, blood cultures and CXR help to rule out infection prior to corticosteroid treatment and establish a baseline prior to therapy. In addition, a baseline CBC helps to place eventual elevation of the white blood cell (WBC) count into context due to demargination and not infection. At our institution, we treat patients for 5 days with 1 g IV solumedrol per day for an initial attack of ATM. In addition to corticosteroid therapy, advances in immune-directed therapy have added an entirely new dimension to what is available for patients affected by ATM. Intravenous immunoglobulin (IVIg) and plasma exchange have been tested in randomized trials and proved to complement corticosteroid therapy, while chemotherapies and monoclonal antibodies are now being incorporated into regimens for patients with refractory or recurrent attacks. With respect to ATM, plasma exchange versus IVIg has been a topic of debate and research; nonetheless, plasma exchange is superior to IVIg in this case for the following reasons. Plasma exchange in a setting of Guillain-Barré syndrome and myasthenia gravis has been shown to deliver immune induction slightly faster than IVIg, with a peak efficacy of IVIg 2-3 weeks after administration. Furthermore, in the author’s opinion, ATM is often antibody mediated as evidenced by the long list of diseases associated with it. Moreover, our ability to identify antibodies is still limited and is demonstrated by the fact that up to one third of ATM is ultimately diagnosed as “idiopathic”. Furthermore, plasma exchange offers a very effective means of initially treating patients with ATM without committing patients to long-term immunosuppression. Due to the risks associated with plasma exchange, most patients should be started on IV corticosteroid therapy, with the decision to add plasma exchange based upon the underlying aetiology, extent of neurologic deficits, and clinical judgment.
Management Principles for Plasma Exchange and IVIg
Here we will discuss some of the finer points of using plasma exchange and IVIg in clinical practice. The dosages and duration of treatment apply to ATM and the remaining diseases discussed in this chapter unless specifically stated otherwise.
Therapeutic plasma exchange was first used to treat myasthenia gravis; since then its application has been extended to treat many disease processes where antibody or suspected antibody-mediated disease occurs.
The actual procedure should be performed in consultation with the institution’s blood bank. Current use of plasma exchange is based on North American and French trials, and although no “optimal” standard of care has been established, general practice still follows these studies. The goal of plasma exchange is to remove the offending IgG and IgM from the bloodstream. Studies have demonstrated that in total, 45% of IgG and 75% of IgM are located in the intravascular space. Plasma exchange every other day will rebalance the antibodies in the intravascular space, which then allows for their removal. Therefore, plasma exchange should involve 250 ml per kilogram of body weight (per exchange) every other day for a total of five exchanges. As a result, based on the kinetics (half-life) of autoantibody resynthesis (5-6 weeks for IgG and 5-6 days for IgM), therapeutic plasma exchange should offer a 1-month window to begin immune modulation if warranted. Plasma exchange itself is a very dynamic process. The fluid removed can be replaced with either albumin solutions or fresh frozen plasma (FFP). The preferred solution is 5% albumin (in saline), which significantly reduces the rate of adverse events but does not confer a therapeutic advantage; and FFP should be avoided unless required to treat a concurrent disease. Due to the need for large bore central venous access, plasma exchange requires special attention to the patient’s baseline coagulation profile and platelet function.
The current and recent medications list should be screened for use of anticoagulants and antiplatelet agents such as Coumadin or Plavix, which may delay initiating plasma exchange. Due to the inherent movement of large volumes of blood during plasma exchange, anticoagulation is needed to prevent clotting. Citrate is the agent of choice and works by binding free ionized calcium from within the blood, preventing it from participating in coagulation. As a consequence, some patients may develop hypocalcaemia.
Patients should be informed that they might experience perioral and peripheral paraesthesias, which are transient and related to the plasma exchange. In addition, electrocardiographic changes including prolongation of the QT interval may also occur as a result of hypocalcaemia. A further consequence of citrate use in plasma exchange is the transient diuresis of electrolytes secondary to the renal excretion of this large cation. Another management point to consider when using plasma exchange is the fluid shifts that occur. As a result of the volume of whole blood removed from the body for exchange, hypotension can be the immediate consequence; and, once the blood is returned with 5% albumin, patients with cardiac or renal insufficiency may develop volume overload due to the oncotic pressure inherent to albumin.
Finally, due to the non-specific nature of exchanging serum plasma with 5% albumin in saline, fibrinogen and clotting factors themselves are lost in the process. This is another reason why plasma exchange occurs every 48 hours to allow for the resynthesis of fibrinogen and clotting factors. When treating a patient with plasma exchange, daily CBC, basic metabolic panel (BMP), coagulation profile and electrolyte levels (ionized calcium, magnesium and phosphate) should be performed and replacement given when needed. Once therapy is complete, removal of the central venous access should be delayed for 48 hrs until the international normalized ratio (INR) falls below 1.4.
IVIg is a pooled product derived from many patients’ plasma; its mechanism of action is not fully understood. In the author’s experience, the following ideas are useful to remember when treating patients with IVIg. When treating diseases discussed in this chapter, unless stated otherwise, utilize a total IVIg dose of 2 g/kg ideal body weight. This dose of 2 g/kg is given in divided doses of 0.4 g/kg per day over 5 days in order to minimize side effects in patients who have never received this medication. If future rounds of IVIg are required, in select patients the total dose of 2 g/kg can be divided over 3 days. When calculating the dosing for your patient, you may land on a dosage that is in between two vial sizes. We recommend to simply round up to the next highest dose. For example, if your calculation equals 32 g/day, round up to the nearest vial, such as a 35 g vial. Second, the clinician should be aware of common side effects (flulike symptoms, headache, hypotension, aseptic meningitis) and potential adverse events (myocardial infarction, stroke, renal failure, venous thrombosis, anaphylaxis) secondary to IVIg administration and discuss these with your patient prior to starting therapy. This will increase both adherence to the protracted time needed to infuse each dose and patient tolerance of side effects.
Subsequently, when ordering baseline serology, also include a serum IgA level to screen for patients with IgA deficiency who may be at risk for anaphylaxis. Many IVIg formulations are available. At our institution we attempt to minimize the risks by using only IgA depleted, sucrose-free IVIg. In addition, running the infusion overnight (6-8 h infusion time) minimizes patient discomfort and being bound to an IV pole. Between each infused dose of IVIg, we suggest continued IV hydration; daily basic serology (BMP, CBC, magnesium, phosphate) should be drawn with attention to changes in renal function, down trending WBC count and platelet levels or changes in electrolyte levels. Finally, 5 days of IVIg infusion and maintenance fluids may cause volume overload in some patients; therefore, concurrent use of diuretics may be warranted in those with cardiac and renal insufficiency.
75.3 Anterior Horn Cell to Neuromuscular Junction
We continue our discussion of neuromuscular weakness, transitioning from the spinal cord to anterior horn cell, peripheral nerve, neuromuscular junction. The topics discussed in this section do not represent all entities that can cause neuromuscular weakness along the peripheral nervous system (PNS); instead we have selected a few topics which are important to understand. It is essential to recognize that neuromuscular weakness in the PNS can present in multiple settings. The emergency room, after treatment of another disease, prolonged hospitalization or intensive care unit (ICU) stay, discovered postoperatively are all clinical settings in which neuromuscular weakness can clinically appear for the first time. Some examples of neuromuscular weakness due to disease of the PNS include:
- One third of patients treated for status asthmaticus.
- Prevalence of up to 46% of patients with a median duration of 7 days in the ICU who were admitted for sepsis, multiorgan failure or prolonged mechanical ventilation.
- Within the group of patients diagnosed with acquired neuromuscular weakness, the specific incidence was 34% for critical illness myopathy (CIM), 35% for critical illness sensorimotor polyneuropathy (CIP), and 30% for combined CIM/CIP in the same patient.
- The risk of acquired neuromuscular weakness is increased with elevated plasma glucose levels.
75.3.1 West Nile Virus
We begin our look at neuromuscular weakness by discussing a viral illness that can affect the anterior horn cell. West Nile Virus (WNV) was first isolated from a woman in Uganda in 1937 and the first case documented in the United States wasn’t until 1999 in New York. Since then, the Centers for Disease Control (CDC) reported over 11,000 cases of this illness and over 1000 deaths as of 2007. WNV is an arthropod-borne and maintained Flavivirus which utilizes mosquitoes as vectors and birds to amplify. Based on data from 2002-2007, the peak incidence of human infection is the summer months of July through September. In addition, cases of West Nile transmission have been reported via organ transplant, blood transfusion, breast milk and transplacentally from mother to foetus. The mechanism of viral entry into the CNS is unknown, but histopathology studies demonstrate the virus has a predilection for neurons.
In the clinical arena, WNV infection leading to neurologic disease is actually quite rare. Data combined from both North America and Europe estimate that up to 70-80% of infections are asymptomatic, 20-30%, develop West Nile Fever (WNF), and only 1% demonstrate neurologic disease or West Nile neuroinvasive disease (WNND). WNF emerges after a 2-14 day incubation period with clinical signs and symptoms similar to the common flu such as headache, myalgias, weakness, fever, nausea and vomiting, diarrhoea and abdominal pain. Patients with WNF can experience lingering symptoms that last weeks to months, but usually recover without sequelae. The 1% who develop WNND can present in three different ways, or in combinations thereof. WNND can present as West Nile encephalitis (WNE), meningitis or flaccid paralysis. About 25-35% of patients will present with pure signs and symptoms of meningitis (fever, nuchal rigidity, photophobia and phonophobia) without encephalitis, whereas 60-75% patients with WNND will present with WNE. This group is characterized as having encephalitis with focal neurologic deficits such as tremor, myoclonus or Parkinsonian features (rigidity and bradykinesia). Furthermore, WNE carries a higher mortality rate of up to 20%, especially with increased age, when compared to WNF and the meningitic form. When the virus attacks motor neurons, the clinical presentation of WNV is an acute, asymmetric flaccid paralysis that begins in one or more limbs. Initially, patients may complain of pain in the limb(s) that subsequently become paralyzed. The weakness can ascend in 50% of cases to include bilateral facial muscles, but the infection should spare sensory modalities. In cases where flaccid paralysis resulted in respiratory neuromuscular weakness, the mortality increases to 50%.
Patients presenting with high fever and meningitis, encephalitis or paralysis should trigger a high index of suspicion for WNV if their history includes spent many hours outdoors during the summer months. CSF evaluation demonstrates pleocytosis, with <500 cells and often lymphocytic in predominance. Furthermore, CSF protein can be elevated and glucose should be normal. Diagnosis is confirmed by IgM antibody to WNV in the CSF if collected within 8 days of illness onset or from serum if collected within 8-14 days of illness onset. While PCR can diagnose diseases such as herpes encephalitis with great accuracy, WNV PCR is positive in only 50% of patients. What can also make arriving at a final diagnosis more difficult is that the serum test of WNV also cross-reacts with other known flavivirus infections such as Japanese encephalitis, St. Louis encephalitis, yellow fever and Dengue. As a result of WNND mimicking other diseases on initial presentation, neuroimaging and electrodiagnostic studies serve to rule out other diseases rather than rule in WNV. If WNND is suspected in the spinal cord, MRI may demonstrate T2-weighted hyperintensity in the anterior (ventral horns) spinal cord. Ultimately, the diagnosis of WNV infection is made on the basis of strong clinical suspicion and diagnostic laboratory testing.
There is no specific treatment for any form of WNV infection and care is only supportive. Recovery from flaccid paralysis does occur, but can take many months and requires aggressive physical rehabilitation.
75.3.2 Guillain-Barré Syndrome
Guillain-Barré Syndrome (GBS) affects one to four persons per 100,000 annually worldwide and two per 100,000 annually in the United States. Most patients who experience GBS ultimately recover; however, up to 20% have permanent disability. Respiratory failure is an initial presentation in 10% of patients with 30% of remaining patients eventually requiring mechanical ventilation. If intubated, the most common causes of morbidity in a GBS patient include systemic infection, venous thromboembolism and cardiac arrhythmia secondary to dysautonomia. Two-thirds of patients, 2 to 4 weeks prior to the onset of neurologic symptoms, will have suffered from an acute illness. Upper respiratory infection, Campylobacter jejuni infection, and CMV are the most common preceding illnesses associated with GBS. In addition, other diseases associated with developing GBS include hepatitis, HIV, herpes virus, influenza, mononucleosis, systemic lupus and lymphoma. Also, when GBS is suspected, inquiring as to any recent vaccinations is also important because epidemiological links have been reported.
Within the first 2 weeks of symptom onset, GBS has certain typical clinical features. Symptoms begin as a progressive ascending weakness from the legs, depressed or absent reflexes and distal extremity paraesthesias. On initial presentation, patients may recall experiencing difficulty with tasks dependant on proximal muscles such as rising from a chair or climbing steps and may complain of low back pain. Acutely, the incidence of weakness is 60%, paraesthesias is 80%, both at the same time is 30%, areflexia is 70% and hyporeflexia is 30%. With time, GBS symptoms tend to evolve with increasing both the areas and magnitude involved, so a typical GBS patient will eventually have weakness in all limbs, areflexia and paraesthesias. As previously stated, weakness in a typical GBS patient is an ascending process beginning in the lower extremities followed by the arms; however, a minority of patients (5-15%) will clinically present with cranial nerve deficits first, then a descending pattern of weakness. Facial and neck weakness will occur in half of patients with ptosis in 5-10%, ophthalmoplegia in 15%, and oropharyngeal weakness in 40%. Diaphragmatic dysfunction is seen in up to 50% of patients, with a combination of both weak shoulder shrug and neck flexion as surrogate markers heralding diaphragmatic weakness and respiratory failure. Sensory symptoms can precede weakness by 1 to 2 weeks, with an early nondescript complaint of generalized malaise and pain (>50%) in the spine. The typical paraesthesia described by patients begins in the fingers as “pins and needles” and is seen first before sensory changes are reported in the distal extremities. As the sensory symptoms evolve, they transition from paraesthesias to a lack of sensation and numbness. Unlike weakness, sensory disturbances in the face and trunk are rare. Autonomic dysfunction, including tachy-brady arrhythmias, orthostatic hypotension, ileus, bladder dysfunction, and abnormal sweating, is seen in up to 50% of patients. Most symptoms will plateau within 2 weeks; rarely, some will take 4 weeks.
The differential diagnosis of a patient who presents with GBS, a GBS variant or a clinically similar picture is very broad and beyond the scope of this chapter, but is listed in Table 75.3.




