Gene (protein)
Chromosome
Inheritance
Role in AD pathogenesis
APP (amyloid precursor protein)
21
Autosomal dominant
Mutations in the APP gene promote cleavage at b or g sites leading to overproduction of Aβ
PSEN1 (presenilin 1)
14
Autosomal dominant
Mutations in the PS1 gene promote cleavage at the g site leading to overproduction of Aβ
PSEN2 (presenilin 2)
1
Autosomal dominant
Mutations in the PS2 gene promote cleavage at the g site leading to overproduction of Aβ
APOE (apolipoprotein E)
19
Modifies age of onset
Promotes the deposition of Aβ?
The most well-established genetic risk factor for late-onset AD is the apolipoprotein E gene (APOE). Three APOE alleles have been identified: e2, e3, and e4. The APOE e4 allele has been linked to the development of AD, whilst epidemiologic as well as pathologic studies have suggested a possible protective effect for the e2 allele. There is an apparent gene-dosing effect according to the actual genotype. One meta-analysis reported the odds of developing AD in heterozygous carriers of the APOE e4 allele as threefold, while the odds for homozygous carriers was almost 15-fold.
The APOE e4 allele operates mainly by decreasing the age of onset with each allele copy lowering the age of onset by almost 10 years. It is therefore a marker of susceptibility rather than a determinative gene. The mechanism whereby APOEe4 influences the development of AD is complex and may be modified by other genes and environmental factors. APOE acts as a cholesterol transporter in the brain, which mediates neuronal protection and repair and is believed to participate in early Aß deposition. The APOE e4 allele has been associated with increased severity of illness including faster cognitive decline, increased risk of conversion from mild cognitive impairment to AD, more neuropsychiatric symptoms, decreased survival time, and increased amyloid load at autopsy. The use of APOE genotype as a diagnostic tool has been examined in several studies but low sensitivity and specificity limit its usefulness and it is not currently recommended for diagnostic use. Table 4.1 summarizes established Alzheimer’s genes and their functional relevance.
Although the APOE e4 allele has been calculated to account for most of the genetic risk in late-onset alzheimer’s disease, a number of candidate genes of smaller effect have also been identified. The advent of large Genome Wide Association Studies (GWAS) has allowed identification of several genes of small effect and findings are collated in the online AlzGene database which allows researchers to compare results and search for consistency between international groups. It is hypothesised that genetic mutations of modest effect in concert with other genetic and environmental factors, may precipitate clinical disease in vulnerable individuals. For example, variant of genes for CLU (clusterin/apolipoprotein J), CR1 (complement receptor 1), and PICALM (phosphatidylinositol-binding clathrin assembly protein) have achieved genome-wide significance in independent GWAS studies. These genetic variants may be involved in reduced clearance of Aß. It is hoped that improved understanding of underlying mechanisms of action may lead to new therapeutic targets to delay the onset and progression of disease.
4.2.2 Lifestyle and Vascular Risk Factors
Although several AD risk factors are genetic in nature, others are determined by environmental or lifestyle influences and may be amenable to modification. Recent years have seen an expansion in the number of epidemiological studies in AD and several risk factors that were traditionally considered as “vascular” have been associated with increased risk of AD (Table 4.2). Longitudinal studies such as the Cardiovascular Risk Factors, Aging, and Dementia (CAIDE) study have found midlife hypertension, hypercholesterolemia, and obesity to be associated with increased risk of dementia and AD in later life.
Table 4.2
Potentially modifiable risk factors found to be associated with Alzheimer’s disease in population studies
Risk factors for Alzheimer’s disease | Protective factors for Alzheimer’s disease |
|---|---|
Hypertension/hypotension | Physical activity |
Obesity | Cognitive and social stimulation |
Diabetes mellitus | Diet (fish consumption > once/week) |
Hypercholesterolemia | Alcohol (light to moderate intake) |
Hyperhomocysteinemia | Higher educational status (>15 year vs. <12 year) |
Stroke | Purpose in life and conscientiousness |
Smoking | Social engagement |
Depression, distress, and loneliness | |
Sleep disturbances |
Clustering of risk factors was observed to increase the risk in an additive fashion. A dementia risk score using data gathered during the CAIDE study predicted dementia with a sensitivity of 0.77, specificity of 0.63, and negative predictive value of 0.98 over 20 years of follow up. This score included variables such as age (≥47 years), low education (<10 years), hypertension, hypercholesterolemia, and obesity. There is now a great deal of interest in developing approaches to help reduce the risk of AD in later life through identifying individuals who might benefit from intensive lifestyle consultations and pharmacological interventions in earlier life. However, not all studies have replicated these findings and one systematic review concluded that the evidence for single clinically defined vascular risk factors was inconsistent at best while the strength of the association was increased by identifying interactions between risk factors such as hypertension and diabetes. In addition, the relationship between cognition and blood pressure is complex with hypertension in midlife and hypotension in later life both associated with increased risk of AD. Although initial case control studies reported a protective effect for smoking, longitudinal studies have now established that smoking is associated with increased risk of AD. Longitudinal studies show that stroke increases the subsequent risk of AD and hyperhomocysteinemia has similarly been reported to increase risk.
Other factors, that have been reported to be protective from population studies, include regular fish consumption, moderate wine intake, and higher educational status. There is also now a significant amount of epidemiological data, which suggests that individuals who are more socially and physically active and engage in more cognitively stimulating activities are at decreased risk of developing dementia and AD. In fact, a recent review concluded that after accounting for non-independence between risk factors, around a third of Alzheimer’s diseases cases worldwide might be attributed to potentially modifiable risk factors. Psychological risk factors are also important. In particular, depression has been found to behave as both a risk factor for and prodromal symptom of AD. Late onset depression may occur as part of the prodrome of early AD but depression, which occurs decades in advance of cognitive decline, is also known to increase risk of AD in a dose-dependent fashion. Psychological distress and loneliness have similarly been reported to increase risk. A number of mechanisms for the association between depression and AD have been proposed. Depression has been independently associated with increased risk of cerebrovascular disease and the chronic neurotoxic effects of elevated glucocorticoids upon hippocampal neurogenesis and repair may also diminish cognitive reserve. Finally, depression may have a more direct neuropathological effect as stress and exogenous glucocorticoids have been shown to increase β-amyloid production in animal models of AD. Having a greater sense of purpose in life and conscientiousness appear to be protective and have both been independently associated with reduced risk of AD. Sleep disturbances, particularly reduced sleep duration, sleep fragmentation, and sleep-disordered breathing, have been implicated as potentially remediable causes of cognitive decline. However, further research is needed to better define the nature of these associations and determine mechanisms which might allow further exploration of preventive and therapeutic strategies. There are already many good reasons why maintaining good psychological health and promoting a physically, cognitively, and socially active lifestyle may be advisable and beneficial for patients.
4.2.3 Other Risk Factors
Data from epidemiological studies reported an association between nonsteroidal anti-inflammatory (NSAID) use and decreased risk of AD while interventional studies in this area have been negative to date. Lipid-lowering medications have similarly been reported to be associated with decreased risk while interventional studies of statins have been largely negative to date. Hormone replacement therapy (HRT) was associated with decreased risk of AD while an interventional study reported an increased risk of dementia, again highlighting the caution with which observational findings must be interpreted. Severe head injury and exposure to toxins such as defoliants and fumigants have been associated with increased risk. Static risk factors include a family history of trisomy 21. Female gender has also been associated with increased prevalence AD. There are many possible reasons for this variable observation although a number of investigators have concluded that it is due to the longer life expectancy in females rather than gender-specific risk factors for the disease.
4.2.4 Pathogenesis
The exact cellular mechanisms leading to neuronal cell death in AD remain uncertain but multiple etiological and pathogenetic hypotheses have been put forward. Macroscopically, the brain in established AD shows a variable degree of cortical atrophy with widening of cerebral sulci and compensatory ventricular enlargement. Microscopically, the disease is characterized by amyloid plaques and neurofibrillary tangles (Fig. 4.1). The current criteria for a pathologic diagnosis of AD require the presence of both amyloid plaques and neurofibrillary tangles in excess of that anticipated for age-matched healthy controls. Amyloid plaques consist of a central core of amyloid protein surrounded by astrocytes, microglia, and dystrophic neurites. Neurofibrillary tangles contain paired helical filaments of abnormally phosphorylated tau protein that occupy the cell body and extend into the dendrites. Neuronal loss or atrophy in the nucleus basalis, locus ceruleus, and raphe nuclei of the brainstem leads to deficits in cholinergic, noradrenergic, and serotonergic transmitters, respectively. The deficit in cholinergic neurotransmission and the observation that this correlated strongly with the degree of cognitive impairment led to the “cholinergic hypothesis” of AD and the subsequent development of cholinesterase inhibitors to redress this deficit.
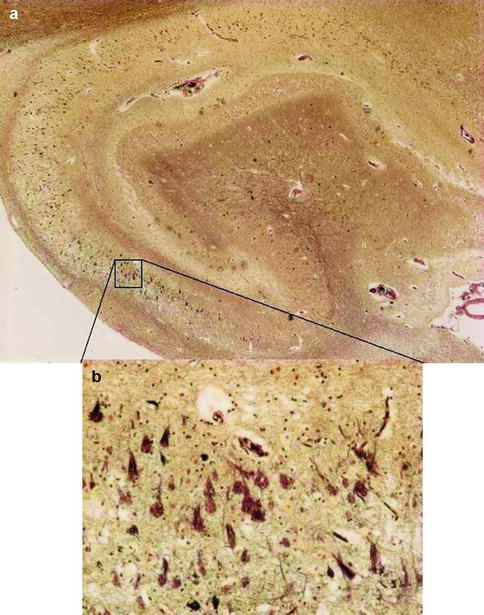
Fig. 4.1
Low power (a) and high power (b) views of hippocampus with neuritic amyloid plaques (P), which consist of a central core of amyloid protein surrounded by astrocytes, microglia, and dystrophic neurites and neurofibrillary tangles (T), which contain paired helical filaments of abnormally phosphorylated tau protein. The current criteria for the histopathological diagnosis of Alzheimer’s disease require the presence of both entities
4.2.5 Amyloid Hypothesis
The amyloid hypothesis remains the best-defined and most studied conceptual framework for AD (Fig. 4.2). Over time, this hypothesis has undergone alterations primarily due to the fact that increased beta amyloid protein (Ab) and plaque formation are no longer considered to be sole triggering factors for deleterious events leading to AD. The exact cellular mechanisms leading to neuronal cell death in AD remain uncertain. The amyloid cascade hypothesis holds that an imbalance between Ab production and clearance plays a critical role in progression of AD. Ab is derived from the much larger transmembrane protein, amyloid precursor protein (APP), by the action of two proteases referred to as beta (b) and gamma (g) secretase. The initial cleavage of APP is mediated by b secretase and then, depending on the exact point of cleavage by g secretase, three principle forms of Ab comprising 38, 40, or 42 amino acid residues, respectively are produced. Most mutations in the APP or presenilin genes alter APP processing resulting in increased levels of Ab. At a certain critical concentration, Ab monomers associate to form neurotoxic oligomers, which then further associate into insoluble fibrils and are deposited as amyloid plaques. The relative amount of Ab 42 formed is important as this longer form of Ab is more prone to aggregate and form oligomers. Ab oligomers could directly inhibit hippocampal long-term potentiation and impair synaptic function in addition to the inflammatory and oxidative stress caused by aggregated and deposited Ab. Much is yet to be learned about the formation and toxicity of these soluble forms of Ab and how they may trigger deleterious changes. The central significance of Ab in the pathogenesis of AD has recently been called into question given negative outcomes from trials of therapeutic agents targeting this pathway. A more current view of the amyloid cascade hypothesis is that other events as well as Ab are important in triggering degenerative processes. The concept of Ab as only one of the factors that causes AD, explains it’s less than perfect correlation with disease severity and it seems increasingly likely that Ab, although necessary, is not by itself, sufficient for AD to occur.
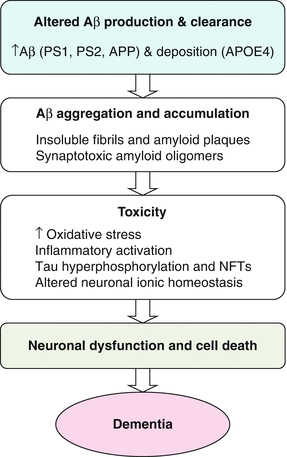
Fig. 4.2
Amyloid cascade hypothesis
4.2.6 Other Proposed Mechanisms
Although amyloid has received much attention with regard to halting the progression of AD, it is not the only target for disease-modifying therapies. Neurofibrillary tangles, which consist of aggregations of hyperphosphorylated tau protein, are another pathologic hallmark of AD. Tau binds to and stabilizes microtubules that are elongated polymers intrinsic to axonal structure and function. When tau is hyperphosphorylated, it aggregates into tangles with resulting destabilization of microtubules and compromised neuronal function. It is unclear whether neurofibrillary tangles are a cause or consequence of AD but their formation may be critical to AD-related cell death. Inflammatory mechanisms have long been known to play an important role in the evolution of AD pathology and many studies have shown a broad variety of inflammatory mediators, including acute phase proteins, cytokines, and chemokines within the vicinity of AD plaques. Neuroinflammation is still considered to be a downstream consequence in the amyloid hypothesis whereby Ab within the CNS brings about activation of microglia, initiating a proinflammatory cascade that results in the release of potentially neurotoxic substances, including cytokines, chemokines, reactive oxygen and nitrogen species, and various proteolytic enzymes, leading to neurodegeneration. It has also been suggested that activation of microglia may lead to phosphorylation of tau and formation of neurofibrillary tangles. However, the exact role of inflammation in the pathology of AD and its mechanisms in terms of the cells involved, which include microglia, astrocytes, and T lymphocytes are still debated.
The frequent co-occurrence of cerebrovascular disease with AD and the fact that fewer neuropathologic lesions of AD appear to result in dementia in the presence of comorbid cerebrovascular disease has been well documented (as per the landmark Nun cohort study). In fact, it is now recognized that mixed pathology is the rule rather than the exception and that cerebrovascular disease is clinically under-recognized and under-reported. The vascular hypothesis of AD goes further and proposes that cerebral hypoperfusion and microvascular pathology may be the primary etiological factor in AD. It proposes that AD develops when two biological events converge; advancing age and the presence of vascular risk factors to create a critically attained threshold of cerebral hypoperfusion (CATCH). This leads to dysregulation of endothelial nitric oxide (NO) production, capillary degeneration, and mitochondrial oxidative stress. The resulting crisis leads to cellular and subcellular pathology involving protein synthesis, development of plaques, inflammatory response, and synaptic damage leading to the manifestations of AD.
4.2.7 Clinical Features
The typical clinical presentation of AD is that of insidious progressive impairment of episodic memory representing early involvement of medial temporal lobe structures with the emergence of additional deficits such as aphasia, apraxia, agnosia, and executive deficits as the disease progresses. Findings from longitudinal studies indicate that neuropsychological deficits in multiple cognitive domains are evident several years in advance of a diagnosis of AD. One meta-analysis reported that the largest deficits in preclinical AD exist in the domains of perceptual speed, executive functioning, and episodic memory with smaller deficits in the domains of verbal ability, visuospatial skills, and attention. This is characterized clinically by initial forgetfulness for daily events with progressive involvement of language skills, decision making, judgment, orientation, recognition, and motor skills. Neuropsychiatric symptoms are frequently observed and occur in 60–98 % of patients with dementia. They are a significant source of distress for patients and families and a major determinant of outcomes such as length of hospital stay and nursing home placement. They ordinarily increase with increasing disease severity but are observed early in the disease process and have been documented in 30–75 % of patients with mild cognitive impairment. Apathy, anxiety, depression, and agitation occur most frequently. Delusions are also common and include themes of theft, intruders, imposters, or other ideas of persecution, reference, or infidelity. Visual and auditory hallucinations are the most common perceptual abnormalities although somatic, olfactory, and tactile hallucinations have also been reported. Functional decline starts with the impairment of higher order (instrumental) functions, such as the management of the house-hold affairs or finances before more gross functions relating to basic self-care are affected. Atypical presentations of AD occur in approximately 6–14 % of cases and are more prevalent in those with younger onset disease. Atypical presentations have been divided into a number of variants including a frontal variant, which may present with executive dysfunction or behavioural symptoms and is difficult to distinguish clinically from frontotemporal dementia. There is also a logopenic variant where there is more prominent impairment of language and a posterior variant where deficits in visuospatial function predominate. These presentations remain relatively uncommon, particularly in late onset disease, where the typical clinical course is one of insidious episodic memory decline with progressive involvement of other cognitive skills as outlined above.
4.2.8 Mild Cognitive Impairment
Neurodegeneration is estimated to start 20–30 years before clinical onset. During this pre-clinical phase, the burden of plaque and tangle pathology gradually increases until the threshold for clinical expression is reached. Mild Cognitive Impairment (MCI) is a clinical classification of patients who manifest cognitive deficits in excess of that expected for normal aging but who do not have significant functional impairment.
The initial diagnostic criteria specified the presence of subjective and objective deficits in memory, with the purpose of detecting patients with the earliest clinical signs of Alzheimer’s pathology for recruitment to clinical trials. Patients in these studies were observed to convert to Alzheimer’s disease at a rate of approximately 10–15 % annually. The diagnostic criteria have since been broadened to include patients with deficits in domains other than memory, to reflect the heterogeneity of both progressive and nonprogressive pathologies represented within this classification. Patients with amnestic deficits in addition to deficits in other domains have the greatest risk of progression to Alzheimer’s disease. Significant variation in rates of conversion to Alzheimer’s disease has been observed depending on the diagnostic criteria used and populations investigated. A recent review of longer-term follow-up studies (5 years or more) indicated that the annual conversion rate of 10–15 % only held true in samples monitored over a short observation period and that the conversion rate was highest shortly after presentation with a marked decline in subsequent years. The authors reported an average cumulative conversion rate of 31.4 % over a mean observation period of 6 years in a sample, which included patients derived from both clinic and community populations. Reported variations in conversion to AD likely reflect variations in the underlying aetiology with conversion rates at the higher end of the range anticipated in individuals with MCI due to AD.
4.2.9 Diagnosis and Revised Diagnostic Criteria
Regarding the diagnosis of Alzheimer’s disease there have been two conceptually overlapping but differing revisions of the National Institute of Neurologic and Communicative Disorders and Stroke – Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria. One developed etc by the International Working Group (IWG) for new Research Criteria for the Diagnosis of Alzheimer’s Disease (AD), the other by the National Institute on Aging – Alzheimer’s Association (NIA-AA) workgroups on diagnostic guidelines for Alzheimer’s disease. Both revisions incorporate biomarkers but differ in how this information is used. Biomarkers are a core requirement in the IWG criteria which are research diagnostic criteria. The NIA-AA revision provides different sets of diagnostic criteria for the different stages of AD, namely the asymptomatic preclinical phase, the symptomatic pre-dementia phase (mild cognitive impairment due to AD), and the dementia phase. The NIA-AA pre-dementia and dementia criteria can be clinically applied even in the absence of biomarkers which are incorporated on a basis of increased or decreased likelihood. Both the NIA-AA criteria and more recently revised IWG criteria (IWG-2) address the issue of atypical presentations of AD which can present as a posterior variant (occipitotemporal or biparietal subtypes), a logopenic (language prominent) variant, or a frontal variant. The IWG-2 criteria outline criteria for diagnosis of typical, atypical and mixed presentations of the disease.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


