Fig. 15.1
Global prevalence of multiple sclerosis- people per 100,000 with MS
However, recent evidence suggests that this latitude gradient in MS incidence may be decreasing. This may be partly due to lifestyle changes leading to decreased sunlight exposure and Vitamin D deficiency. Another possible explanation is the so-called hygiene hypothesis. It is thought that exposure to infections in early childhood has a protective effect against the later development of autoimmune diseases. As socioeconomic factors lead to decreased childhood infections, thus autoimmunity in adulthood may be increasing over time. It has long been known that MS is about twice as common in women compared to men. Similarly however, it has recently been reported that the female- to- male ratio of MS is increasing over time. One study reported the female-to-male ratio increased from approximately 2 for patients diagnosed in 1940 to 4 for those diagnosed in 2000. The cause of the observed rise in this ratio remains unclear though increased smoking in women during the twentieth century is one putative contributing factor. Though MS is thought to affect Caucasians more than other racial groups, a recent US study found that the risk of developing MS was higher in black women when compared to white women though the risk did not differ between men of various ethnicities.
15.1.2 Clinical Course and Subtypes of Disease
Multiple Sclerosis can develop in many forms, the most common of which is relapsing remitting MS (RRMS), characterised by discrete attacks or relapses followed by remissions. A relapse represents the development of a new neurological symptom or worsening of a pre-existing symptom that persists for greater than 24 h and is not associated with infection. Relapses are usually followed by a period of complete or incomplete remission of variable duration. MS is characterised by this relapsing-remitting onset in about 85 % of cases. When a patient initially presents with a first relapse and does not yet meet criteria for MS, this is said to be a clinically isolated syndrome (CIS).
Eventually, remissions are of shorter duration, relapse associated disability accumulates, and the patient enters a progressive disabling phase. About 80 % of relapsing remitting cases will progress to secondary progressive MS (SPMS) within two decades. The secondary progressive stage is characterised by a slow disease progression as irreversible disability is gradually accrued. Its onset is usually determined retrospectively. The mean time interval for conversion from RRMS to SPMS is 10.4 years. Relapses persist in around 40 % of cases during the progressive phase. The primary progressive MS subtype (PPMS) is present in 15–20 % of MS cases and is characterised by at least 1 year of relapse free progression at onset. There are no remissions as the patient suffers a progressive decline in function. A slowly developing motor onset is more typical of PPMS however it can also be characterised by progressive ataxia. Between 15 and 20 % of MS patients will prove to have a benign form of multiple sclerosis, remaining fully functional in all neurological systems more than 10 years after disease onset. Acute MS, or Marburg’s disease, is thankfully very rare. It is characterised by rapid disease progression and an exceptionally severe course usually leading to death within a few years of onset.
15.1.3 Clinical Features
MS is a clinically heterogenous disease and almost any neurological symptom can occur during the course of the illness. The symptoms and signs of multiple sclerosis merely reflect the functional anatomy of impaired conduction due to demyelination in the optic nerves, brain, and spinal cord. A number of clinical presentations are very typical of MS. A specific and characteristic sensory symptom seen in MS is the “useless hand syndrome” where one hand is functionally impaired due to proprioceptive loss. Acute bilateral internuclear opthalmoplegia (INO), Lhermitte’s symptom, Uthoff’s phenomenon, unilateral facial myokimia, bilateral trigeminal neuralgia, and unilateral remitting optic neuritis, are all also suggestive of MS when occurring in young adults. Among the commonest chronic symptoms of the disease are fatigue and depression, bladder irritative problems and urinary retention, limb weakness and spasticity, sensory symptoms, neuropathic pain, and cognitive impairment. Paroxysmal symptoms such as trigeminal neuralgia and tonic spasms can also frequently occur.
15.2 Pathology and Aetiology of MS
15.2.1 Pathology
The pathological hallmark of MS is demyelination. It is caused by inflammation targeting the myelin sheath and is associated with the formation of focal lesions within the CNS known as plaques. The classic acute MS plaque is characterised by perivascular infiltration of activated macrophages and T lymphocytes. The pathology of the acute plaque is heterogenous and four different patterns have been described based on myelin protein loss, the location and extension of the plaque, the pattern of oligodendrocyte destruction, and the involvement of complement components. The pattern may not be static within a given individual, and more than one pattern can be observed in the same patient. Partial remyelination of plaques occurs to a variable extent. Severe demyelination results in axonal loss and damage to surrounding oligodendrocytes.
The classical view of MS as a disease that exclusively affects the white matter has more recently been refuted. Pathological studies and modern MRI modalities have convincingly demonstrated diffuse pathology in both the normal appearing white matter (NAWM) of the brain and spinal cord, as well as cortical demyelination affecting the grey matter in MS patients. This diffuse pathology is seen even at very early stages of disease.
MS is believed to start with activation and proliferation of autoreactive CD4+ T helper lymphocytes in the periphery. These cells then migrate across the blood brain barrier (BBB), recognise auto-antigens, and initiate inflammation via the production of pro-inflammatory cytokines. This inflammatory cascade cumulates in damage to the myelin sheath, oligodendrocytes, and axons. It is an extremely complex process involving multiple cell types, cytokines, and co-stimulatory molecules. In the progressive stage of the disease less active inflammation is observed and disability is thought to occur due to degenerative changes. Nonetheless, oligodendrocyte progenitor cells (OPCs) capable of remyelination have been observed in the white matter plaques of chronic MS patients suggesting that remyelination may be possible even in the late stages of disease. This represents an encouraging therapeutic target for treatment of progressive MS.
15.2.2 Genetics of MS
MS is not inherited in a Mendelian fashion and is likely to be polygenic. There is however a familial recurrence rate of about 15 %. The age-adjusted risk is higher for siblings (3 %), parents (2 %), and children (2 %) than for second degree and third-degree relatives. Recurrence in monozygotic twins is around 35 %. Linkage studies in multiplex families have confirmed that variation within the major histocompatibility complex (MHC) has the greatest individual effect on risk. A large genome wide association study (GWAS) has recently replicated this finding and has identified at least another 29 susceptibility loci. Immunologically relevant genes are significantly overrepresented among those mapping close to these loci confirming that MS is primarily an immune mediated disease.
15.2.3 Environmental Factors and MS
Migration studies, geographical variation in disease rates, and high rates of discordancy in identical twins suggest that environmental agents must play a role in the pathogenesis of MS. The role of Vitamin D status in MS has gained importance in recent years as both epidemiological and experimental data increasingly suggest that Vitamin D deficiency plays an importance role in both MS pathogenesis and disease activity. A case control study involving seven million US military personnel with stored serum found that lower risk of MS was associated with higher levels of 25-hydroxyvitamin D. This effect was interestingly seen in white people but not in black or Hispanic people. A study that included 42,0000 people found that significantly fewer people with MS are born in November and significantly more are born in May, a fact that is putatively linked to the Vitamin D status of the mother during gestation. It has been shown that Vitamin D levels are significantly lower in relapsing remitting MS patients compared to controls and are also lower in patients experiencing disease activity compared to those in remission. A recent study demonstrated that a 50-nmol/L (20-ng/mL) increment in average serum 25-hydroxyvitamin D levels within the first 12 months of disease predicted a lower relapse rate, a lower rate of T2 lesion accumulation on MRI, and a lower rate of yearly brain atrophy from months 12–60. This suggests that vitamin D deficiency is a strong predictor of future relapse activity and disease progression.
With respect to lifestyle factors only smoking has emerged as a moderate risk factor for MS. More recently childhood obesity has been highlighted as a possible risk factor for later development of MS. Over the years several transmissible agents have been implicated as possible causes of MS though to date no infectious trigger has been definitively identified. In recent times the leading candidates include Epstein-Barr virus (EBV) and Human Immunodeficiency Virus (HIV). The epidemiological data associated EBV infection with MS remains strong. We know that almost all patients with MS (>99 %) have evidence of prior EBV infection when compared to only about 90 % of controls. People who have had symptomatic EBV infection (infectious mononucleosis) are at increased risk of developing MS for up to 30 years after the original infection. In this large Danish cohort the ratio of observed: expected MS cases was 2.27. High titres of EBV antibodies predict a higher risk of MS than low titres and Hodgkin’s lymphoma, another EBV associated condition, occurs more frequently in MS patients when compared to controls. None of these facts provide proof of causation but they do suggest a role for EBV in the pathogenesis of MS.
Though both MS and HIV are widely documented in the medical literature, only one case of co-existent MS and HIV infection has been documented. The individual in question was commenced on combination anti-retroviral therapy (cART) following which their MS remained quiescent for up to 12 years of follow up. It was postulated that the anti retroviral therapy may be coincidentally treating or preventing MS or that perhaps the HIV infection itself exerted some form of biological protection. This case report prompted further enquiry into this phenomenon. A large UK linkage study investigating the possible association between MS and HIV was recently reported. The relative risk of MS in people with HIV, relative to those without HIV, was 0.38. The magnitude of this effect (>60 %) is at the highest level of any prognostic risk factor reported to date. This finding may prompt future trials of anti-retroviral therapies in MS patients.
15.3 Current Diagnostic Criteria
The diagnosis of MS relies on providing evidence for dissemination of CNS lesions in both time and space, as well as excluding alternative diagnoses. MS is primarily a clinical diagnosis and there is no one test that is pathognomonic of the disease. However, the widespread availability of magnetic resonance imaging (MRI) has revolutionized the diagnostic workup and has led to earlier diagnosis of disease than ever before. Typical MRI findings in MS patients include ovoid lesions located perpendicularly to the corpus callosum (Dawson’s fingers), in the juxtacortical white matter, in the cerebellum, brainstem, and cervico-thoracic spinal cord (see Fig. 15.2). Fluid attenuated inversion recovery (FLAIR) is the most sensitive imaging sequence for these white matter abnormalities. Active plaques that are associated with disruption of the blood brain barrier enhance with administration of paramagnetic contrast agents such as gadolinium.
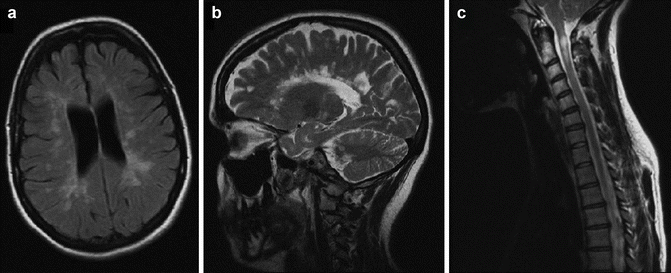
Fig. 15.2
MRI images typical of MS reproduced with patient permission. (a) Axial FLAIR image showing ovoid periventricular and juxta-cortical white matter hyper intensities, (b) sagittal T2 image showing “Dawson’s fingers” perpendicular to the corpus callosum, and (c) a sagittal T2 spinal image demonstrating an MS plaque in the cervical cord
The McDonald criteria are a set of diagnostic criteria for MS that incorporate the clinical characteristics of the disease alone or in combination with MRI features. These criteria were first introduced in 2001 by an international panel and were revised in 2005 and most recently in 2010 (see Table 15.1). To demonstrate dissemination of lesions in space (DIS) using MRI, one or more T2 lesions must be seen in areas typical of MS; namely the periventricular, juxtacortical or infratentorial white matter or within the spinal cord. Dissemination of lesions in time (DIT) can be demonstrated using MRI when a new T2 and/or gadolinium enhancing lesion develops relative to a baseline MRI scan. Alternatively, the simultaneous presence of asymptomatic gadolinium enhancing and non-enhancing lesions at any time is sufficient to demonstrate DIT.
Table 15.1
2010 revised McDonald criteria for the diagnosis of multiple sclerosis
Clinical presentation | Additional information needed to make MS diagnosis |
|---|---|
2 or more relapses Objective clinical evidence of 2 or more lesions with history of prior relapse | None, clinical evidence will suffice. MRI brain is still desirable |
2 or more relapses Objective clinical evidence of 1 lesion | Dissemination in space demonstrated by MRI or await further relapse implicating a different CNS site |
1 relapse Objective clinical evidence of 1 lesion | Dissemination in space demonstrated by MRI or await 2nd relapse implicating a different CNS site And Dissemination in time demonstrated by MRI or a 2nd clinical relapse |
Insidious neurological progression suggestive of primary progressive MS | One year of disease progression and dissemination in space demonstrated by 2 of the following: 1 or more T2 brain lesions typical of MS 2 or more T2 lesions in the spinal cord Positive CSF- OCBs and /or raised IgG index |
Relapse- a new neurological symptom lasting at least 24 h in the absence of infection. There must be at least 30 days between onset of one relapse and onset of another | CSF- cerebrospinal fluid OCB- oligoclonal band |
Examination of the cerebrospinal fluid (CSF) is also a useful ancillary investigation in suspected multiple sclerosis cases. In MS intrathecal synthesis of oligoclonal immunoglobulin G (IgG) bands are found in approximately 90 % of patients. A simultaneous blood sample is required to demonstrate the intra-thecal origin of the bands as the passive transfer of bands from the systemic circulation is of no diagnostic value. Isoelectric focusing and immunodetection of oligoclonal bands (OCBs) is the gold standard technique used to identify intrathecal antibody synthesis. A raised IgG index is also suggestive of MS but is not as sensitive or specific as the presence of OCBs.
15.4 Differential Diagnosis and Syndromes That Overlap with MS
15.4.1 Neuromyelitis Optica (NMO)
NMO (Devic’s disease) is an inflammatory demyelinating disease of the CNS that for many years was thought to be a variant of MS but is now known to be immunologically and pathologically distinct. Clinically it presents with optic neuritis and myelitis, often with a poor recovery and a relapsing course. MR imaging typically shows longitudinally extensive cord lesions, extending over three or more vertebral segments. Brain lesions can be clinically silent and are seen in about 60 % of patients often in the diencephalon and hypothalamus. Pathologically NMO is characterised by demyelination, neuronal loss, and often pronounced necrosis. The discovery of aquaporin-4 autoantibodies in 2004 (AQP4-Ab, also known as NMO-IgG) in the serum of patients with NMO represented a considerable advance in both the diagnosis and treatment of the disease and distinguished NMO from MS as an independent disease entity. The specificity of these antibodies is between 90 and 100 % in the correct clinical context. More recently, antibodies to myelin oligodendrocyte glycoprotein (MOG) have been reported in AQP4-Ab negative cases with a typical NMO phenotype. However, the exact diagnostic implications of this finding are still under investigation.
More women than men have NMO (ratio 9:1) and the median age of onset is more than 10 years older than in MS. Moderate pleocytosis in the CSF of NMO patients is seen more commonly than in MS and OCBs are only seen in about 30 % of cases. In NMO the CSF OCB status can change over time, which is uncommon in MS. More recently high concentrations of IL-6 have been found in the CSF of NMO patients which may help in differentiating it from other demyelinating diseases. Therapy should be initiated early in NMO and the therapeutic pathway is different from that of MS. Azathioprine and Rituximab are recommended as first line agents. Other immunosuppressant agents such as methotrexate, mycophenylate mofetil, and mitoxantrone are recommended as second line agents. Promising new therapies such as anti- AQP4-Ab and anti-IL-6 receptor biologicals are emerging.
AQP4-Ab have also been demonstrated in patients with clinical syndromes atypical of NMO such as brainstem syndromes including intractable vomiting, narcolepsy, neuroendocrine disturbances, and olfactory dysfunction. The term “NMO spectrum disorders” is often used as an umbrella term for these varied presentations.
15.4.2 Acute Disseminated Encephalomyeltis (ADEM)
ADEM is an inflammatory demyelinating disorder that can often be considered in the differential diagnosis of an initial demyelinating event or clinically isolated syndrome (CIS). It can present with a number of clinical symptoms but usually includes encephalopathy as well as multifocal symptoms suggestive of an inflammatory disorder. ADEM differs from MS and NMO in that it is a monophasic illness that does not relapse and therefore does not require disease modifying therapy (DMT). A recent review has proposed a number of clinical, imaging, and laboratory features that are helpful in distinguishing ADEM from a first presentation of other inflammatory disorders. ADEM is seen much more commonly in the paediatric population but can also occur in adults. Unlike MS, it is often preceded by an infection or vaccination. It should be considered when there is a multifocal polysymptomatic presentation, encephalopathy, seizures, fever, headache, and bilateral optic neuritis. On CSF examination a lymphocytic pleocytosis is more commonly seen in ADEM than in MS and CSF OCBs are seen less frequently than in MS and often resolve when present in ADEM. MRI will often demonstrate large (1–2 cm) multifocal areas of T2 abnormality in the CNS white matter often with diffuse enhancement and grey matter involvement in addition. Periventricular involvement is less prominent than in MS. A brain biopsy is sometimes warranted when the diagnosis remains uncertain and the pathological hallmark of ADEM is perivenular inflammation with “sleeves of demyelination”. Intravenous methylprednisolone is the most common treatment used in practice when an infectious aetiology has been excluded, IVIG may be an effective alternative.
15.4.3 Differential Diagnosis of MS
Diagnostic criteria for MS emphasize that that an alternative explanation for the presentation must be considered and excluded before a diagnosis of MS can be made. International consensus based guidelines have been published to guide the clinical, laboratory, and imaging assessments of patients with possible MS in order to best exclude alternative diagnoses. The differential diagnosis of MS is wide and includes diseases that can mimic both relapsing and progressive neurological disturbances. These are outlined in Table 15.2 although such lists are always selective and incomplete.
Table 15.2
Differential diagnoses of progressive and relapsing forms of multiple sclerosis
Differential diagnosis of a progressive cord syndrome | Differential diagnosis of a relapsing CNS disorder |
|---|---|
Tumour Intramedullary- glioma, ependymoma Extramedullary- meningioma, neurofibroma Extradural- metastasis Vascular- Dural arteriovenous malformation Metabolic B12 deficiency Copper deficiency Vitamin E deficiency Phenyketonuria Degenerative– Amyotrophic lateral sclerosis Infectious HIV HTLV-1 Syphilis Tuberculosis Schistosomiasis Inflammatory Neurosarcoidosis Systemic lupus erythematosus Paraneoplastic Genetic Hereditary spastic paraparesis Freidreich’s ataxia Adrenomyeloneuropathy Toxic- Nitrous oxide | Vascular disease CADASIL Cerebral amyloid angiopathy Antiphospholipid antibody syndrome Fabry’s disease Vasculitis Behcet’s disease Neurosarcoidosis Sjogren’s syndrome Susac’s syndrome Sysyemic lupus erythematosus Primary angiitis of the CNS Mitochondrial disease MELAS Chronic infection Lyme disease Neurosyphilis HTLV-1 HIV encephalitis Fungal/parasitic infections Whipple’s disease CADASIL- Cerebral Autosomal Dominant Arteriopathy with subcortical infarcts and leucoencephalopathy MELAS- Mitochondrial Encephalopathy with Lactic Acidosis and Stroke-like episodes |
15.4.4 Disease Modifying Therapies (DMTs)
In the long-term management of MS, a number of disease modifying therapies (DMTs) are used to reduce the relapse rate and therefore reduce the amount of relapse- associated disability accumulated during the relapsing remitting phase of the illness. Several new DMTs have been developed in recent years many of which are more targeted and efficacious than their predecessors. Each of these agents have undesirable side effects and the risk benefit ratio of each therapy has to be considered for each individual patient. There is currently no DMT licensed for use in progressive MS and this remains a critical area of unmet need in MS therapeutics. In RRMS it is now recognised that there is a therapeutic “window of opportunity” during the first 5 years of the disease during which aggressive control of the inflammatory phase of the illness is of most benefit in the long term. It remains unclear to date whether early suppression of relapses and neuro-inflammation with any drug will ultimately prevent the onset of progressive disease.
15.4.5 “First- Generation” Self Injectable Therapies
The first DMTs to be licensed for use in RRMS were self administered injections given either subcutaneously or intramuscularly at a variable frequency depending on the specific agent used. The type 1 beta-interferons were the first immunomodulatory agents to be licensed for use in MS in 1993, and while they are used routinely in RRMS patients, their precise mechanism of action remains to be elucidated. There are currently five commercially available beta-interferon preparations in clinical use (see Table 15.3). Glatiramer acetate (GA, Copaxone) is a synthetic peptide composed of four amino acids designed specifically to mimic the structure of myelin basic protein (MBP). MBP is the principle component of CNS myelin and is therefore a putative autoantigen in MS. Glatiramer acetate received US Food and Drug Administration (FDA) approval for use in MS patents in 1987. All these drugs have comparable efficacy, reducing the relapse rate by about a third, reducing the number of new brain lesions in RRMS patients, and slowing but not preventing the onset of disease progression. They have all established long-term safety but are limited by injection-related skin reactions, and flu-like side effects seen with the interferon-beta preparations. A PEGylated form of interferon-beta 1a plegridy with a longer half-life and a reduced dosing frequency (once every 2 weeks) has recently received a license in RRMS.
Table 15.3
First- generation self-injectable disease modifying therapies for relapsing remitting MS
Drug | Interferon-beta 1b | Interferon-beta 1b | Interferon-beta 1a | Interferon-beta 1a | PegInterferon-beta 1a | Glatiramer acetate |
|---|---|---|---|---|---|---|
Brand name | Betaferon | Extavia | Avonex | Rebif | Plegridy | Copaxone |
Year approved | 1993 | 2009 | 1996 | 2002 | 2015 | 1997 |
Dose | 250 μg | 250 μg | 30 μg | 22 or 44 μg | 125 μg | 20 mg or 40 mg |
Route | SC | SC | IM | SC | SC | SC |
Frequency | Every other day | Every other day | Once/week | 3 times/week | once every 2 weeks | Daily or All days (40 mg) (20 mg) |
15.4.6 Oral DMTS
Three oral agents have been approved for use in relapsing MS: fingolimod, teriflunomide and dimethyl-fumarate (BG-12). Efficacy data from pivotal randomised controlled trials of these agents are summarized in Table 15.4
Table 15.4
Summary of efficacy data from pivotal controlled trials for recently developed disease modifying therapies for relapsing MS
Drug | Fingolimod | Dimethyl fumarate (BG-12) | Teriflunomide | Natalizumab | Alemtuzumab |
|---|---|---|---|---|---|
Brand Name | Gilenya | Tecfidera | Aubagio | Tysabri | Lemtrada |
Year approved | 2010 | 2013 | 2012 | 2004, 2006 | 2014 |
Dose | 0.5 mg | 240 mg | 7 mg or 14 mg | 300 mg | 12 mg |
Route | PO | PO | PO | IV | IV |
Frequency | Daily | Twice daily | Daily | Once every 4 weeks | Annually 5 doses year 1 3 doses year 2 and thereafter as required |
Clinical relapse Relative RR | 54 % | 51 % | 31 % | 68 % | 55 % (vs rebif 44)a 49 % (vs rebif 44)b |
Disability progression Relative RR | 30 % | 38 % | 26 % (14 mg dose only) | 42 % | NSa 42 % (vs rebif 44)b
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
