Fig. 1
Cataplectic attacks in Doberman pinschers. Emotional excitations, appetizing food or playing, easily elicit multiple cataplectic attacks in these animals. Most cataplexy attacks are bilateral (97.9 %). Atonia initiated partially in the hind legs (79.8 %), front legs (7.8 %) neck/face (6.2 %), or whole body/complete attacks (6.2 %) Progression of attacks was also seen (49 % of all attacks) (Fujiki et al. 2002)
Cataplexy is an extremely variable clinical symptom (Gelb et al. 1994). Most often, it is mild and occurs as a simple buckling of the knees, head dropping, facial muscle flickering, sagging of the jaw or weakness in the arms. Slurred speech or mutism is also frequently associated. It is often imperceptible to the observer and may even be only a subjective feeling difficult to describe, such as a feeling of warmth or that somehow time is suspended (Wilson 1927; Gelb et al. 1994). In other cases, it escalates to actual episodes of muscle paralysis that may last up to a few minutes. Falls and injury are rare and most often the patient will have time to find support or will sit down while the attack is occurring. Long episodes occasionally blend into sleep and may be associated with hypnagogic hallucinations. Children with narcolepsy often (about 1/3 of patients) present with a previously unrecognized description of cataplexy that we coined “cataplectic facies,” consisting of a state of semipermanent eyelid and jaw weakness (Serra et al. 2008).
Patients may also experience “status cataplecticus”. This rare manifestation of narcolepsy is characterized by subintrant cataplexy that lasts several hours per day and confines the subject to bed. It can occur spontaneously or more often upon withdrawal from anticataplectic drugs (Parkes et al. 1975; Passouant 1970; Hishikawa 1995).
Cataplexy often improves with advancing age. In rare cases it disappears completely but in most patients it is better controlled (probably after the patient has learned to control their emotions) (Billiard et al. 1983; Rosenthal et al. 1990).
2.3 Sleep Paralysis
Sleep paralysis is present in 20–50 % of all narcoleptic subjects (Roth 1980; Hishikawa 1976; Parkes et al. 1974; Yoss and Daly 1960). It is often associated with hypnagogic hallucinations. Sleep paralysis is best described as a brief inability to perform voluntary movements at the onset of sleep, upon awakening during the night, or in the morning. Contrary to simple fatigue or locomotion inhibition, the patient is unable to perform even a small movement, such as lifting a finger. Sleep paralysis may last a few minutes and is often finally interrupted by noise or other external stimuli. The symptom is occasionally bothersome in narcoleptic subjects, especially when associated with frightening hallucinations (Rosenthal 1939).
Whereas excessive daytime sleepiness and cataplexy are the cardinal symptoms of narcolepsy, sleep paralysis occurs frequently as an isolated phenomenon, affecting 5–40 % of the general population (Dahlitz and Parkes 1993; Fukuda et al. 1987; Goode 1962). Occasional episodes of sleep paralysis are often seen in adolescence and after sleep deprivation, thus prevalence is high for single episodes.
2.4 Hypnagogic and Hypnopompic Hallucinations
Abnormal visual (most often) or auditory perceptions that occur while falling asleep (hypnagogic) or upon waking up (hypnopompic) are frequently observed in narcoleptic subjects (Ribstein 1976). These hallucinations are often unpleasant and are typically associated with a feeling of fear or threat (Hishikawa 1976; Rosenthal 1939). Polygraphic studies indicate that these hallucinations occur most often during REM sleep (Hishikawa 1976; Chetrit et al. 1994). These episodes are often difficult to distinguish from nightmares or unpleasant dreams, which also occur frequently in narcolepsy.
Hypnagogic hallucinations are most often associated with sleep attacks and their content is described by the patient. The hallucinations are most often complex, vivid, dream-like experiences (“half sleep” hallucinations) and may follow episodes of cataplexy or sleep paralysis, a feature that is not uncommon in severely affected patients. These hallucinations are usually easy to distinguish from hallucinations observed in schizophrenia or related psychotic conditions.
Compared to cataplexy, hypnagogic and hypnopompic hallucinations themselves do not have specificity for the diagnosis of narcolepsy, as up to 20–30 % of normal people exhibit these symptoms in various situations (Fukuda et al. 1987; Takeuchi et al. 1992). The folklore over the world describes similar phenomenon, including “Kanashibari” in Japan, literally meaning “bound or fastened in metal”, “Pinyin” in China, literally meaning” ghost pressing on body”.
2.5 Other Important Symptoms
One of the most frequently associated symptoms is insomnia, best characterized as a difficulty to maintain nighttime sleep. Typically, narcoleptic patients fall asleep easily, only to wake up after a short nap unable to fall asleep again before an hour or so. Narcoleptic patients do not usually sleep more than normal individuals over the 24 h cycle but frequently have a very disrupted nighttime sleep (Hishikawa et al. 1976; Broughton et al. 1988; Montplaisir et al. 1978). This symptom often develops later in life and can be very disabling.
Frequently associated problems are periodic leg movements (Godbout 1985; Mosko et al. 1984), REM behavior disorder, other parasomnias (Mayer et al. 1993; Schenck and Mahowald 1992), and obstructive sleep apnea (Mosko et al. 1984; Chokroverty 1986.
Narcolepsy was reported to be associated with changes in energy homeostasis several decades ago. Narcolepsy patients are frequently (1) obese (Honda et al. 1986; Schuld et al. 2000), (2) more often have insulin-resistant diabetes mellitus (Honda et al. 1986), (3) exhibit reduced food intake (Lammers et al. 1996) and (4) have lower blood pressure and temperature (Mayer et al. 1997; Sachs and Kaisjer 1980). These findings however, had not received much attention since they were believed to be secondary to sleepiness or inactivity during the daytime. More recently however, it was shown that these metabolic changes may be found more specifically in hypocretin deficient patients (Nishino et al. 2001; Kok et al. 2003; Hara et al. 2001), suggesting a direct pathophysiological link.
Narcolepsy is a very incapacitating disease. It interferes with every aspect of life. The negative social impact of narcolepsy has been extensively studied. Patients experience impairments in driving and there is a high prevalence of either car or machine-related accidents. Narcolepsy also interferes with professional performance, leading to unemployment, frequent changes of employment, working disability or early retirement (Broughton et al. 1981; Aldrich 1989; Alaila 1992). Several subjects also develop symptoms of depression, although these symptoms are often masked by anticataplectic medications (Broughton 1976; Broughton et al. 1981; Roth and Nevsimalova 1975).
3 The Prevalence of Narcolepsy
The prevalence of narcolepsy has been investigated in several ethnic groups and countries. One of the most sophisticated prevalence studies was a Finnish cohort study consisting of 11,354 twin individuals (Hublin et al. 1994). All subjects who responded to a questionnaire with answers suggestive of narcolepsy were contacted by telephone. Clinical interviews were performed and polysomnographic recordings were then conducted in five subjects considered to be narcoleptic. Sleep monitoring finally identified three narcoleptic subjects with cataplexy, thus leading to a prevalence of 0.026 % (Hublin et al. 1994). All 3 subjects were dizygotic twins and the co-twins were not affected (Hublin et al. 1994). Other prevalence studies have led to similar prevalence values (0.02–0.067 %) in Great Britain (Ohayon et al. 1996), France (Ondzé et al. 1999), the Czech Republic (Roth 1980), 5 European countries (Ohayon et al. 2002) and in the United States (Dement et al. 1973; Silber et al. 2002).
A study performed in 1945 in African American navy recruits also led to 0.02 % in this ethnic group for narcolepsy-cataplexy (Solomon 1945). Narcolepsy-cataplexy may be more frequent in Japan and less frequent in Israel. Two population-based prevalence studies led to a 0.16 and 0.18 % prevalence figure in Japan (Honda 1979; Tashiro 1994). However, these studies used only questionnaires and interviews but not polysomnography to confirm the diagnosis. In Israel, only a few narcoleptic patients have been identified when compared to the large population of subjects recruited into sleep clinics (Lavie and Peled 1987). This has led to the suggestion that the prevalence of narcolepsy could be as low as 0.002 % in this ethnic group.
The age of onset varies, from early childhood to the fifties, with two peaks, a larger one that occurs at around 15 years of age and a smaller peak at approximately 36 years of age (Dauvilliers et al. 2001). Similar results were found in two different populations but the reasons for this bimodal distribution remains obscure. Incidence of the disease was reported to be 1.37/100.000 per year (1.72 for men and 1.05 for women) in Olmsted County in Minnesota, and the incidence rate was highest in the second decade, followed in descending order by the 3rd, 4th and 1st decade (Silber et al. 2002).
4 Genetic and Environmental Factors in Nacrolepsy
A familial tendency for narcolepsy has been recognized since its description in the late 19th century (Westphal 1877). Starting in the 1940s, several studies were published that investigated the familial history of small cohorts of narcoleptic probands (Yoss and Daly 1960; Krabbe and Magnussen 1942; Nevsimalova-Bruhova and Roth 1972; Kessler et al. 1979; Baraitser and Parkes 1978; Honda et al. 1983). Using standard diagnostic criteria, more recent studies have showed rates of familial cases as 4.3 % in Japan (Honda 1988), 6 % in the USA (Guilleminault et al. 1989), 7.6 % in France, and 9.9 % in Canada (Dauvilliers et al. 2001). In addition to subjects who fulfill all diagnostic criteria for narcolepsy–cataplexy, other relatives may report isolated recurrent sleep episodes and unexplained EDS; these subjects may suffer from an incomplete and milder form of the disorder (Billiard et al 1994). Studies also revealed that the risk of a first-degree relative developing narcolepsy–cataplexy is 1–2.0 %, 10–40 times higher than the risk in the general population (Billiard et al. 1994; Mignot 1998; Hublin et al. 1994).
A limited number of twin cases studies have been published. Of 16 monozygotic (MZ) twin pairs with at least one affected twin, only four (or five depending on the criteria used for concordance) were concordant (Mignot 1998). Although genetic predisposition is likely to be involved in the development of narcolepsy, the relatively low rate of concordance in narcoleptic MZ twins indicates that environmental factors also play a role in the development of the disorder. The nature of the possible environmental factors involved is not known. Frequently cited factors are head trauma (Lankford et al. 1994), sudden change in sleep/wake habits (Orellana et al. 1994), and various infections (Roth 1980). Although these factors may be involved, there are no documented studies demonstrating increased frequency when compared to multiple control groups.
5 Neurobiology of Wakefulness
In order to understand the neurobiology of hypersomnia, we will first discuss current understandings of the neurobiology of wakefulness. Sleep/wake is a complex physiology regulated by brain activity, and multiple neurotransmitter systems such as monoamines, acetylcholine, excitatory and inhibitory amino acids, peptides, purines, and neuronal and non-neuronal humoral modulators (i.e., cytokines and prostaglandins) (Nishino and Kotorii 2009; Jones 2005) are likely to be involved. Monoamines are perhaps the first neurotransmitters recognized to be involved in wakefulness (Nishino 2009; Jouvet 1972) and the monoaminergic systems have been the most common pharmacological targets for wake-promoting compounds in the past years. On the other hand, prototypical hypnotics target the gammaaminobutyric acid (GABA)-ergic system, a main inhibitory neurotransmitter system in the brain (Nishino and Mignot 1997; Nishino 2004).
Cholinergic neurons also play critical roles in cortical activation during wakefulness (and during REM sleep) (Nishino 2009; Jones 2005). Brainstem cholinergic neurons originating from the laterodorsal and pedunculopontine tegmental nuclei activate thalamocortical signaling, and cerebral cortical activation is further reinforced by direct cholinergic projections from the basal forebrain. However, currently no cholinergic compounds are used in sleep medicine, perhaps due to the complex nature of the systems and prominent peripheral side effects.
Monoaminergic neurons, such as norepinephrine (NE) containing locus coeruleus neurons, serotonin (5-HT) containing raphe neurons, and histamine containing tuberomammillary neurons (TMN) are wake-active and act directly on cortical and subcortical regions to promote wakefulness (Nishino and Kotorii 2009; Jones 2005). In contrast to the focus on these wake-active monoaminergic systems, researchers have often underestimated the importance of dopamine (DA) in promoting wakefulness. Most likely, this is because the firing rates of midbrain DA-producing neurons (ventral tegmental area [VTA] and substantia nigra) do not have an obvious variation according to behavioral states (Steinfels et al. 1983). In addition, DA is produced by many different cell groups (Björklund and Lindvall 1984), and which of these promote wakefulness remains undetermined. Nevertheless, DA release is greatest during wakefulness (Trulson 1985), and DA neurons increase discharge and tend to fire bursts of action potentials in association with significant sensory stimulation, purposive movement, or behavioral arousal (Ljungberg et al. 1992). Lesions that include the dopaminergic neurons of the VTA reduce behavioral arousal (Jones et al. 1973). Furthermore, recent work has also identified a small wake-active population of dopamine-producing neurons in the ventral periaqueductal grey that project to other arousal regions (Lu et al. 2006). People with DA deficiency from Parkinson’s disease are often sleepy (Moller et al. 2000), and dopamine antagonists are frequently sedating. These physiologic and clinical evidences clearly demonstrate that DA also plays a role in wakefulness.
Wakefulness (and various physiological systems associated with wakefulness) is essential for the survival of creatures and thus is likely to be regulated by multiple systems, each may have a distinct but a complementary role. Some arousal systems may have essential roles for cortical activation, attention, cognition, or neuroplasticity during wakefulness while others may only be active during specific times to promote particular physiology during wakefulness. Some of the examples may be motivated-behavioral wakefulness or wakefulness in emergency states. Wakefulness may thus likely be maintained by many systems with differential roles to be fully alert. Similarly, the wake-promoting mechanism of some drugs may not be able to be explained by a single neurotransmitter system.
6 Sleep Physiology and Narcolepsy Symptomes
Since narcolepsy is a prototypical EDS disorder and since the major pathophysiology of narcolepsy-cataplexy (i.e. deficient in hypocretin neurotransmission) has recently been revealed, the discussion of neurophysiological aspects of narcolepsy will help understand neurobiology of EDS and cataplexy, a pathognomonic symptom of hypocretin deficient narcolepsy.
Narcolepsy patients manifest symptoms specifically related to dysregulation of REM sleep (Nishino and Mignot 1997). In the structured, cyclic process of normal sleep, two distinct states—REM and 3 stages (S1, S2, S3) of non-REM (NREM) sleep—alternate sequentially every 90–110 min in a cycle repeating 4–5 times per night (Nishino et al. 2004). As EEG signals in humans indicate, NREM sleep, characterized by slow oscillation in thalamocortical neurons (detected as cortical slow waves) and muscle tonus reduction, precedes REM sleep, when complete muscle atonia occurs. Slow wave NREM predominates during the early phase of normal sleep, followed by a predominance of REM during the later phase (Nishino et al. 2004).
Notably, sleep and wake are highly fragmented in narcolepsy, and affected subjects could not maintain long bouts of wake and sleep. Normal sleep physiology is currently understood as dependent upon coordination of the interactions of facilitating sleep centers and inhibiting arousal centers in the brain, such that stable sleep and wake states are maintained for specific durations (Nishino et al. 2004). An ascending arousal pathway, running from the rostal pons and through the midbrain reticular formation, promotes wakefulness (Nishino et al. 2004a, b; Saper et al. 2005). As discussed earlier, this arousal pathway may be composed of neurotransmitters (acetylcholine, NE, DA, excitatory amino acids), produced by brainstem and hypothalamic neurons (hypocretin/orexin, and histamine) and also be linked to muscle tonus control during sleep (Nishino et al. 2004; Saper et al. 2005). Whereas full alertness and cortical activation require coordination of these arousal networks, effective sleep requires suppression of arousal by the hypothalamus (Saper et al. 2005). Narcolepsy patients may experience major neurological malfunction of this control system originated in the hypothalamus.
Narcoleptics (both type 1 and type 2) exhibit a phenomenon, termed short REM sleep latency or sleep onset REM period (SOREMP), in which they enter REM sleep more immediately upon falling asleep than normal (Nishino and Mignot 1997). In some cases, NREM sleep is completely bypassed and the transition to REM sleep occurs instantly (Nishino and Mignot 1997). SOREMS are not observed in idiopathic hypersomnia, suggesting a distinct etiology from narcolepsy.
Moreover, intrusion of REM sleep into wakefulness may explain cataplexy, sleep paralysis, and hypnogogic hallucinations, which are symptoms of narcolepsy. Although sleep paralysis and hallucinations manifest in other sleep disorders (sleep apnea syndromes and disturbed sleep patterns in the normal population) (Aldrich et al. 1997) (see also “sleep paralysis and hallucinations section) (Aldrich et al. 1997), cataplexy is pathognomic for narcolepsy (Nishino and Mignot 1997). As such, identifying cataplexy’s unique pathophysiological mechanism emerged to be potentially crucial in understanding overall pathophysiology of narcolepsy.
7 Discovery of Hypocretin Ligand Deficiency in Human Narcolepsy
The significant roles, first, of hypocretin deficiency and, subsequently, of postnatal cell death of hypocretin neurons as the major pathophysiological process underlying narcolepsy with cataplexy, were established from a decade of investigation in, employing both animal and human models. In 1998, the simultaneous discovery of a novel hypothalamic peptide neurotransmitter by two independent research groups proved pivotal (Sakurai et al. 1998; De Lecea et al. 1998). One group called the peptides “hypocretin” because of their primary hypothalamic localization and similarities with the hormone “secretin” (De Lecea et al. 1998). The other group called the molecule “orexin” (after the meaning of appetite in Greek) after observing that central administration of these peptides increased appetite in rats (Sakurai et al. 1998). These neurotransmitters are produced exclusively by thousands of neurons, which are localized in the lateral hypothalamus, and project broadly to specific cerebral regions and more densely to others (Peyron et al. 1998).
Within a year, Stanford researchers, using positional cloning of a naturally-occuring familial canine narcolepsy model, identified an autosomal recessive mutation of hypocretin receptor 2 (Hcrtr 2) responsible for canine narcolepsy, characterized by cataplexy, reduced sleep latency, and SOREMPs (Lin et al. 1999). This finding coincided with the simultaneous observation of the narcolepsy phenotype, characterized by cataplectic behavior and sleep fragmentation, in hypocretin ligand deficient mice (prepro-orexin gene knockout mice) (Chemelli et al. 1999). Together, these findings confirmed hypocretins as principal sleep/wake-modulating neurotransmitters and prompted investigation of the hypocretin system’s involvement in human narcolepsy.
Although screening of high risk patients with cataplexy (i.e., familial, early onset and/or HLA negative cases) did not reveal hypocretin-related gene mutation as a major cause of human narcolepsy, narcoleptic patients did exhibit low CSF hypocretin-1 levels (Nishino et al. 2000) (Fig. 2). Post-mortem brain tissue of narcoleptic patients, assessed with immunochemistry, radioimmunological peptide assays, and in situ hybridization, revealed hypocretin peptide loss and undetectable levels of hypocretin peptides or prepro-hypocretin RNA (Fig. 2). Further, melanin-concentrating hormone (MCH) neurons, which are normally located in the same brain region (Peyron et al. 2000), were observed intact, thus indicating that damage to hypocretin neurons and its production is selective in narcolepsy, rather than due to generalized neuronal degeneration.
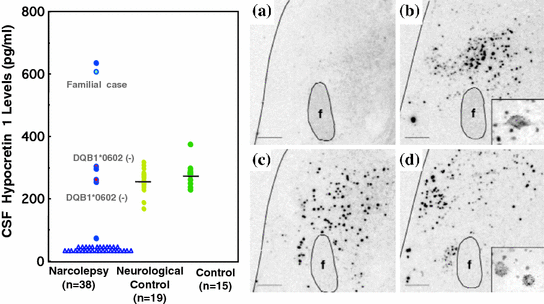
Fig. 2
Hypocretin deficiency in narcoleptic subjects. a CSF hypocretin-1 levels are undetectably low in most narcoleptic subjects (84.2 %). Note that two HLA DqB1*0602-negative and one familial case have normal or high CSF hypocretin-1 levels. b Preprohypocretin transcripts are detected in the hypothalamus of control (b) but not in narcoleptic subjects (a). Melanin-concentrating hormone (MCH) transcripts are detected in the same region in both control (d) and narcoleptic (c) sections. f and fx, fornix. Scale bar represents 10 mm (a–d) [from Peyron et al. (2000)]
As a result of these findings, a diagnostic test for narcolepsy, based on clinical measurement of CSF hypocretin-1 levels for detecting hypocretin ligand deficiency, became available (ICSD-2 2005). Whereas CSF hypocretin-1 concentrations above 200 pg/ml almost always occur in controls and patients with other sleep and neurological disorders, concentrations below 110 pg/ml are 94 % predictive of narcolepsy with cataplexy (Mignot et al. 2002) (Fig. 3). As this represents a more specific assessment than the multiple sleep latency test (MSLT), CSF hypocretin-1 levels below 110 pg/ml are indicated in the ICSD-2 (2005) as diagnostic of narcolepsy with cataplexy (ICSD-2 2005).
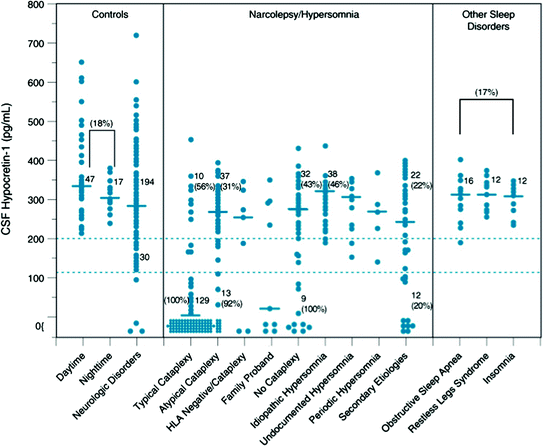
Fig. 3
CSF hypocretin-1 levels in individuals across various control and sleep disorders. Each point represents the crude concentration of Hypocretin-1 in a single person. The cutoffs for normal (>200 pg/mL) and low (<110 pg/mL) Hypocretin-1 concentrations are shown. Also noted is the total number of subjects in each range, and the percentage human leukocyte antigen (HLA)-DQB1*0602 positivity for a given group in a given range is parenthetically noted for certain disorders. Note that control carrier frequencies for DQB1*0602 are 17 to 22 % in healthy control subjects and secondary narcolepsy, consistent with control values reported in whites. In other patient groups, values are higher, with almost all Hypocretin-deficient narcolepsy being HLA DQB1*0602 positive. The median value in each group is shown as a horizontal bar. [Updated from previously published data in (82)] (Mignot et al. 2002)
Moreover, separate coding of “narcolepsy with cataplexy” and “narcolepsy without cataplexy” in the ICSD-2 underscores how discovery of specific diagnostic criteria now informs our understanding of narcolepsy’s nosology; narcolepsy with cataplexy, as indicated by low CSF hypocretin-1, appears etiologically homogeneous and distinct from most narcolepsy without cataplexy cases, exhibiting normal hypocretin-1 levels (Mignot et al. 2002). In the 3rd revision, narcolepsy was reclassified as hypocretin deficient/Type 1 and hypocretin non-deficient/Type 1 narcolepsy, and this classification is solely based on the pathophysiological findings. The potential of hypocretin receptor agonists (or cell transplantation) in narcolepsy treatment is currently being explored, and identifying hypocretin deficient status may be useful in identifying appropriate patients as candidates for a novel therapeutic option, namely hypocretin replacement therapy.
Soon after the discovery of human hypocretin deficiency, researchers identified specific substances and genes, such as dynorphin and neuronal activity-regulated pentraxin (NARP) (Crocker et al. 2005) and, most recently, insulin-like growth factor binding protein 3 (IGF BP3) (Honda et al. 2009), which colocalize in neurons containing hypocretin. These findings underscored selective hypocretin cell death as the cause of hypocretin deficiency (as opposed to transcription/biosynthesis or hypocretin peptide processing problems) because these substances are also deficient in postmortem brain in lateral hypothalamic area (LHA) of hypocretin deficient narcoleptic patients (Crocker et al. 2005; Honda et al. 2009). Furthermore, these findings, in view of the generally late onsets of sporadic narcolepsy compared with those of familial cases, suggest that postnatal cell death of hypocretin neurons constitutes the major pathophysiological process in human narcolepsy with cataplexy.
A large kindred of familial narcolepsy (12 affected members) has been reported in Spain (Hor et al. 2011). Affected members do not exhibit any symptoms suggesting symptomatic cases of narcolepsy, and were diagnosed as familial diopathic narcolepsy-cataplexy. The family includes a pair of dizygotic twins concordant for narcolepsy-cataplexy in the third generation; the distribution of the disorder indicates an autosomal-dominant transmission of the disease-causing gene. Hor et al. recently performed linkage analysis and sequenced coding regions of the genome (exome sequencing) of three affected members with narcolepsy and cataplexy, and identified a missense mutation in the second exon of oligodendrocyte glycoprotein MOG (Hor et al. 2011). A c.398C > G mutation was present in all affected family members but absent in unaffected members and 775 unrelated control subjects (Hor et al. 2011). Affected members were hypocretin deficient, but association with HLA DQB1*0602 was not observed (Hor et al. 2011). The mutation may induce secondary hypocretin deficiency with or without immune mediated mechanisms. MOG has been linked to various neuropsychiatric disorders and is considered a key autoantigen in multiple sclerosis and in its animal model, experimental autoimmune encephalitis (Clements et al. 2003), and thus autoimmune mechanisms may also be involved in these cases. However, even if autoimmune mechanisms are involved in these cases, it is possible that the primary target for the immune attack is not the hypocretin system. These results suggest the heterogeneity of the etiology of idiopathic narcolepsy-cataplexy.
8 What Causes Loss of Hypocretin Neurons?
Because of its strong association with certain human leukocyte antigen (HLA) alleles (i.e., HLA DQB1*0602 and DQA1*0102) (Mignot et al. 1997), it has long been speculated that narcolepsy results from an autoimmune-mediated mechanism (see Table 1). Increase in Antistreptolysin O [ASO] titer in narcolepsy, especially in the period close to the disease onset, was reported (Aran et al. 2009). However, attempts to identify specific autoantibodies have been unsuccessful for decades. Recently, Tribbles homolog 2 (Trib2) was reported as a candidate antigen involved in the destruction of hypocretin neurons in narcolepsy (Cvetkovic-Lopes et al. 2010). Trib2 was shown to be abundantly expressed in hypocretin neurons, and levels of Trib2-specific antibodies were much higher in patients with narcolepsy, especially shortly after the disease onset. Thus, Trib2 is the first antibody specifically associated with hypocretin deficient narcolepsy (Cvetkovic-Lopes et al. 2010). However, it is still unknown if Trib2-specific antibodies are directly involved in cell death, or if the antibody production is a consequence of cell damage by other unknown mechanisms (Lim and Scammell 2010).
 Hypocretin Story
Hypocretin Story
 Models of Narcolepsy
Models of Narcolepsy
 Induced Modulation of REM Sleep and Its Loss Associated Patho-Physiological Changes Are Mediated Through Locus Coeruleus
Induced Modulation of REM Sleep and Its Loss Associated Patho-Physiological Changes Are Mediated Through Locus Coeruleus
 and Circadian Influences in Sleep and Psychiatric Disorders: A Review of Experimental and Computational Modelling Studies
and Circadian Influences in Sleep and Psychiatric Disorders: A Review of Experimental and Computational Modelling Studies
 of Thalamocortical Pathways by Orexins
of Thalamocortical Pathways by Orexins
 Narcolepsy or Hypersomnia, with and Without Orexin (Hypocretin) Deficiency
Narcolepsy or Hypersomnia, with and Without Orexin (Hypocretin) Deficiency




Table 1
Clinical characteristics Type 1 and Type 2 narcolepsy, and idiopathic hypersomnia
Daytime sleepiness duration awaken-refreshed | Other symptoms | MSLT | HLADQB1*0602 positivity | Low CSF hypocretin levels (<110 pg/ml) | ||
|---|---|---|---|---|---|---|
Sleep latency | SOREMPS | |||||
Narcolepsy type 1 | ||||||
Narcolepsy-cataplexy | Short (<30 min) (+) | Cataplexy REM sleep-related symptoms | <8 mina | ≥2 | >90 % | 85−90 > 90 % in HLA positive |
Narcolepsy type2 | ||||||
Narcolepsy w/o cataplexy | Short (<30 min) (+) | Cataplexy (−) REM sleep-related symptoms | <8 mina | ≥2 | 40–50 % | 10–20 % (almost all HLA positive) |
Secondary narcolepsy (due to the medical condition) | Varied (±) | with and without Cataplexy REM sleep-related symptoms | <8 mina | ≥2 | Not systematically studied | Most cases studiedb |
Idiopathic hypersomnia with long sleep time | Long (>30 min) (−) | Cataplexy (−) prolonged nighttime sleep (≥ 10 h) autonomic nervous dysfunction | <8 mina | ≤1 | No consistent results | Normal |
Idiopathic hypersomnia without long sleep time | Varied (−) | Cataplexy (−) no prolonged nighttime sleep (<10 h) autonomic nervous dysfunction | <8 mina | ≤1 | No consistent results | Normal |
aLess than 8 min is considered for both narcolepsy and idiopathic hypersomnia in the 2nd and 3rd revision of ICSD
bCSF hypocretin-1 measures wee done only in a limited number of secondary narcolepsy. Although many cases associated EDS with and without cataplexy exhibited moderately reduced CF hypocretin-1 levels, cases with sleep abnormalities/cataplexy are habitually selected for CSF hypocretin-1 measures, and it is not known if all or a large majority of cases with low CSF hypocretin-1 levels with CNS interventions, exhibit EDS/cataplexy. CSF lumbar sac cerebrospinal fluid; hcrt hypocretin; HLA human leukocyte antigen; MSLT multiple sleep latency test; SOREMP sleep-onset rapid eye movement period
Related posts:
Hypocretin Story
Models of Narcolepsy
Induced Modulation of REM Sleep and Its Loss Associated Patho-Physiological Changes Are Mediated Through Locus Coeruleus
and Circadian Influences in Sleep and Psychiatric Disorders: A Review of Experimental and Computational Modelling Studies
Narcolepsy or Hypersomnia, with and Without Orexin (Hypocretin) Deficiency
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
