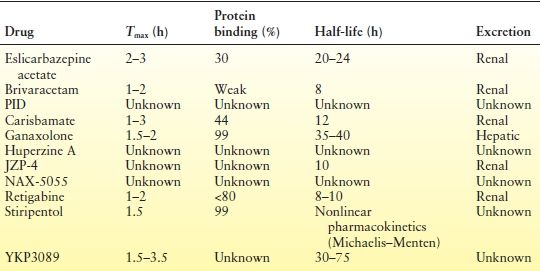
Table 68.2 Important Pharmacokinetic Parameters of New Antiepileptic Compounds in Humans
In the fifth edition of this textbook, this chapter discussed 11 anticonvulsant drugs (11). Information on brivaracetam, carisbamate, ganaxolone, stiripentol, huperzine A, and YKP3089 has been updated. Nax 5055, JZP-4, and propylisopropyl acetate (PID) will not be discussed in this chapter as the interval development of these agents has stalled or ceased. Compounds newly developed as potential AEDs since the writing of the last edition have been added.
The chapter has also been reorganized; drugs whose chemical structures are related to preexisting parent compounds are discussed first under the derivative compound section, followed by neurosteroids (NS), anti-inflammatory agents, and other structurally novel compounds.
DERIVATIVE COMPOUNDS
Levetiracetam Derivatives: Brivaracetam
Brivaracetam (UCB 34714) is a pyrrolidone derivative in the same class as levetiracetam and piracetam. Like levetiracetam, it binds to the synaptic vesicle protein 2A (SV2A), but with higher affinity. In contrast to levetiracetam, brivaracetam also inhibits sodium channel currents as demonstrated in rat cortical neurons in vitro (12). It exhibits superior activity against secondarily generalized motor seizures in corneally kindled mice and prevents clonic convulsions in audiogenic-susceptible mice (13). Brivaracetam suppresses spike-and-wave discharges in Generalized Absence Epilepsy Rat from Strasbourg (GAERS) rats and also suppresses both motor seizure severity and after-discharge duration in amygdala-kindled rats (13). Brivaracetam was developed with the hope that it would (i) be more potent than levetiracetam due to its higher affinity binding, (ii) provide benefit to a larger percent of patients because of its additional impact on sodium channels, or (iii) have a more favorable behavioral side effect profile. None of these potential benefits have yet been proven.
Brivaracetam is rapidly absorbed following oral administration, displays linear pharmacokinetics, binds weakly to plasma proteins, and has an approximately 8-hour half-life (14,15). Inactive metabolites are primarily renally cleared (15). In contrast to levetiracetam, brivaracetam has been demonstrated in vitro to have some impact on metabolizing enzymes; it inhibits epoxide hydrolase and to a lesser extent CYP3A4 and 2C19 and is a weak inducer of CYP3A4 (16). Brivaracetam slightly reduces carbamazepine concentrations, while slightly increasing levels of carbamazepine-10,11-epoxide. Brivaracetam has equivocal effects on phenytoin, possibly lowering serum concentrations, and has no effect on the concentrations of lamotrigine, levetiracetam, oxcarbazepine, topiramate, and valproic acid (6,17). High doses of brivaracetam have been shown to moderately reduce the estrogen and progesterone components of oral contraceptives, but this has not been shown to impact ovulation (6,16). However, a dose of 100 mg/day did not modify the pharmacokinetics of oral contraceptive steroid components (30 g ethinyl estradiol and 150 g levonorgestrel) (18). Enzyme-inducing AEDs increase brivaracetam clearance, leading to a 30% reduction of AUC (7). There is no change in exposure in elderly and renally impaired patients, indicating that no dose adjustments should be necessary (19). However, severe hepatic impairment will increase exposure by about 50% (7,18).
Tolerability is good overall; the majority of treatment- emergent adverse events (TEAEs) reported in Phase III trials were mild to moderate (8), the most common being headache, somnolence, dizziness, and fatigue (8,20). Cardiac repolarization in healthy subjects is not affected with doses of 75 and 400 mg BID (21).
Three Phase III clinical trials (Study N01252, N01253, and N01254) utilizing the primary efficacy end point of percent reduction in baseline-adjusted partial-onset seizure frequency have been conducted. Patients were randomized to a variety of drug doses (20, 50, and 100 mg/day in Study N01252; 5, 20, and 50 mg/day in Study N01253; and 20 mg/day with variable stepwise increases up to a maximum of 150 mg/day in Study N01254). In Study N01253, statistical significance was reached for the primary efficacy end point with the 50 mg/day dose versus placebo (P = 0.02) (22); this was not achieved for the 50 mg dose in Study N01252, but was at 100 mg/day dosing (P = 0.05) (22). In clinical trial N01254 (23), median percent reduction from baseline in partial-onset seizure frequency/week was 26.9% for brivaracetam versus 18.9% for placebo (P = 0.070), while 50% responder rates were 30.3% for brivaracetam versus 16.7% for placebo (P = 0.006) (23). Preliminary data also suggest possible efficacy of brivaracetam as an add-on agent in adults with generalized epilepsy (consisting of a combination of idiopathic, cryptogenic, symptomatic, and unknown, with 34/36 patients completing the treatment period) (23). The median generalized seizure days/week decreased to 0.63 on brivaracetam compared to placebo (1.26); median percentage reductions from baseline and ≥50% responder rates in generalized seizure days/week also favored brivaracetam treatment (42.6% and 44.4%, respectively) over placebo (20.7% and 15.4%, respectively) (23).
Multiple additional Phase III studies as well as multicenter open-label trials are ongoing at the time of writing of this chapter, including studies in children.
Vigabatrin Derivatives: CPP-115
Vigabatrin increases levels of γ-aminobutyric acid (GABA) in the brain by inhibiting its degradation through inactivation of the enzyme GABA-aminotransferase (GABA-AT) (24). Derivatives of vigabatrin have been created; strengthening the molecular structure of the drug has led to the development of a more potent analog with stronger binding capacity to the target enzyme GABA-AT (25). The molecule (1S,3S)-3-amino-4-difluoromethylenecyclopentanecarboxylic acid, a conformationally rigid vigabatrin analog, is a potent inactivator of GABA-AT and approximately 186 times more potent than is S-vigabatrin in vitro (25). However, the latter molecule possesses relatively low lipophilicity, which could limit permeability across the blood–brain barrier (25). The tetrazole analog (1S,3S)-3-amino-4-difluoromethylenyl-cyclopentanoic acid (CPP-115) is more highly lipophilic (25). In addition, CPP-115 is a time- and concentration-dependent inhibitor of GABA-AT (25), less potent than is its parent compound, but more potent in vitro than is vigabatrin (25). CPP-115 was over 100 times more effective in the treatment of infantile spasms in a rat model, and efficacy against drug addiction in rat models was also demonstrated (26). And importantly, much less retinal damage has been observed at high doses of CPP-115 compared to effective doses of vigabatrin (26). CPP-115 has been licensed to Catalyst Pharmaceutical Partners, Inc., which has begun Phase I clinical trials at the time of writing of this chapter.
NEUROSTEROIDS
Étienne-Émile Baulieu (27) first defined NS as steroids synthesized de novo in the nervous system from cholesterol independent of the peripheral endocrine glands. This definition was expanded by Paul and Purdy (28) who further characterized “neuroactive steroids” as including “natural or synthetic steroids” that alter neuronal excitability. GABAA receptors are primary NS targets; at low concentrations, NS potentiate the action of GABA, and at higher concentrations, they directly activate the receptor at sites distinct from benzodiazepine and barbiturate modulatory sites (29), and anticonvulsant tolerance to NS has not been seen (30,31). NS exhibit broad-spectrum anticonvulsant effects in animal models, showing efficacy against Pentylene Tetrazole (PTZ), bicuculline, pilocarpine, and kindling-induced seizures (32). Similar to other GABA agents, however, NS may exacerbate generalized absence seizures (33,34). Ganaxolone is the only synthetic NS-based compound currently in development and is discussed below.
GANAXOLONE
Ganaxolone (3α-hydroxy-3β-methyl-5α-pregnan-20-one) is the 3-β-methyl analog of the NS allopregnanolone (7). It is a positive allosteric modulator of the GABAA receptor and is similar to its natural analog allopregnanolone in potency and efficacy (35). Ganaxolone has protective antiepileptic activity in multiple seizure models (35–38). It is highly protein bound (>99%) (7), is rapidly absorbed, and is primarily hepatically metabolized via CYP3A4/3A5 enzymes (7). In vitro studies have failed to show significant interactions between ganaxolone and other AEDs (39). Ganaxolone is not a CYP3A4 inhibitor and has shown low or no potential to induce CYP3A4 metabolism, although concomitantly administered strong CYP3A4 inducers (carbamazepine and phenytoin) can increase ganaxolone’s clearance (8). Ganaxolone has a 10-hour effective half-life and a terminal elimination half-life of 35 to 40 hours (7). Metabolites are eliminated in urine (25%) and feces (69%) (32). Plasma values are 5 to 15 times higher when taken with food, compared to fasting values; thus, several unique formulations have been created including two solid capsule forms, one immediate-release and the other pH-sensitive delayed-release. Ganaxolone possesses a favorable safety and toxicity profile in animals and is safe and well tolerated in humans (8). The most common TEAEs are headache, dizziness, fatigue, and somnolence (8,40). The 3β-methyl component of ganaxolone eliminates back conversion to the hormonally active intermediate dihydroprogesterone, potentially avoiding hormonal side effects (41).
In one Phase II clinical trial, 1500 mg/day of ganaxolone resulted in an 18% decrease of mean weekly seizure frequency, compared with a 2% increase in the placebo group over a 10-week treatment period (P = 0.014) (8). Twenty-six percent of subjects treated with ganaxolone versus 13% of those treated with placebo (P = 0.057) experienced >50% reduction in seizures during the maintenance phase (8). Sustained efficacy was demonstrated in a 104-week open-label extension of a 10-week double-blind randomized study, with no new safety concerns identified (8). Three Phase II open-label, adjunctive therapy studies have been performed in the pediatric population. The largest enrolled a total of 45 patients with partial or generalized refractory seizures, ages 2 to 15 years. A maximum dose of 12 mg/kg three times daily was administered for an 8-week maintenance period. Twenty-seven patients (60%) completed the entire study and 12 patients (27%) experienced a >50% reduction in seizure frequency (39). Ganaxolone is currently in Phase III development for epilepsy (8).
ANTI-INFLAMMATORY AGENTS
Targeting hyperexcitability and inflammation is another potential approach to seizure control (42). Evidence suggests that glia-mediated inflammation plays a role in the pathogenesis of seizures and epilepsy; one proposed mechanism is glial effects on the IL1β cascade, which ultimately leads to NMDA-mediated enhancement of Ca2+ influx, which in turn promotes excitability and excitotoxicity (43,44). Using therapies that target the inflammatory cascade particularly for those patients severely resistant to standard AED treatment may be helpful; results from animal studies are promising (45).
HE3286 (Triolex)
The orally active HE3286 is a synthetic derivative of the anti-inflammatory dehydroepiandrosterone (DHEA) metabolite, androstene-3β,7β,17β-triol (βAET) (46). While anti- inflammatory, HE3286 is not immunosuppressive (47); it is thought to have an anti-inflammatory mechanism of action distinct from corticosteroids and other agents (48). HE3286 has been used to treat collagen-induced arthritis, where it has been associated with various immune-related changes including the reduction of proinflammatory cytokines tumor necrosis factor-α, IL-6, IL-1β, and IL-23 (48).
Thus far, HE3286 demonstrates low potential for direct or indirect toxicity and possesses suitable pharmacokinetics for clinical development (47). It possesses a terminal half-life slightly longer in males than in females (T1/2: 8.2 and 5.4 hours, respectively) (47). HE3286 readily permeates the blood–brain barrier in mice (47). It is extensively metabolized, and some of the various metabolites have been found to interact with CYP enzymes (47). Pharmacokinetic evaluations are ongoing. Toxicology studies in rats revealed no systemic or neurotoxicity at daily doses of up to 400 mg/kg in 4-week studies; doses of 200 mg/kg and 30 mg/kg in 13- and 26-week studies, respectively, also resulted in no toxicity (47). Dose-dependent, generally mild, estrogenic effects were observed in both sexes (47).
At the time of writing of this chapter, HE3286 was in preclinical development for use in refractory epilepsy using a chronic seizure mouse model at the Mario Negri Institute for Pharmacological Research, Milan, Italy.
VX-765
VX-765 ((S)-1-((S)-2-{(1-(4-amino-3-chloro-phenyl)-methanoyl)-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide) is a selective and reversible inhibitor of interleukin converting enzyme, also known as caspase 1 (8,49). By this mechanism, it is able to reduce the production of IL-1β, which is thought to inhibit neuronal hyperexcitability.
Two small Phase IIA clinical trials have been completed. The first was a 6-week trial in which subjects were randomized 4:1 to VX-765 900 mg three times daily (TID) (n = 48), or placebo (n = 12). There was no long-term extension phase, but patients continued to count seizures for 6 weeks after drug was discontinued. The most common side effect was dizziness, and no serious side effects attributable to VX-765 emerged. 18.8% of subjects in the VX-765 group had a 50% reduction in seizures versus 8.3% in the placebo group (P = NS). However, in a post hoc analysis, seizure frequency appeared to diminish further in a “hybrid” period consisting of the last 2 weeks of treatment and the first 2 weeks of the posttreatment period, consistent with a delayed onset of treatment seen in animal studies (8,50). Subsequently, a 13-week dose-ranging study was initiated. Patients were randomized (1:1:1:1:1) before the first dose of study drug on day 1 to receive placebo or VX-765 300 mg, 600 mg, 900 mg, or 1200 mg TID. The study was initially designed to enroll 500 patients. It was stopped administratively due to a decision by the company not to pursue development, when only 55 patients had been randomized. At present, the fate of the molecule is uncertain (51).
OTHER STRUCTURALLY NOVEL COMPOUNDS
2-Deoxy-d-Glucose
2-Deoxy-d-glucose was discovered during studies exploring the possible molecular mechanisms underlying the anticonvulsant effect of the ketogenic diet (8). 2-Deoxy-d-glucose is a nonmetabolizable glucose analog that inhibits glycolysis and that has been shown to have beneficial acute and chronic antiepileptic effects in a number of in vitro and rat models of seizures (52). A recent report suggests that it may exert its anticonvulsant properties by up-regulation of the potassium receptor subunits Kir6.1 and Kir6.2 (53). There have been some concerns about its safety in animal models with report of cardiac muscle vacuolization and increased mortality in rats (54). The significance and reversibility of these findings are currently under investigation. An intriguing aspect of this compound is that it may be preferentially taken up at the site of active seizure focus and thus may have a specific role in the treatment of acute repetitive seizures and status epilepticus. Phase 11 studies in humans have been planned, including studies of use in status epilepticus.
Cannabinoids
There is a lot of current interest in the medical use of marijuana and its constituent compounds for the treatment of epilepsy. There are hundreds of unique compounds in marijuana (cannabis), and the ones that are unique to the plant are called cannabinoids. Two neuroactive compounds in marijuana (cannabis) are tetrahydrocannabinol (Δ9-tetrahydro-cannabinol (Δ9-THC), a psychoactive compound) and cannabidiol (non-psychoactive). Sativex, a derivative containing both constituents, has recently been approved in a number of countries for the treatment of pain and spasticity in multiple sclerosis (55). In the day-to-day practice of epileptology, some patients have reported that the use of marijuana has beneficial effects on seizure control, and indeed, the use of cannabis has a long history of use for the control of seizures in human epilepsy. Both THC and cannabidiol have been tested in animal models of epilepsy, but recently, there has been more focus on cannabidiol. Notably, while THC engages the endocannabinoid system, cannabidiol does not. However, cannabidiol does engage a number of other targets and is considered to be antioxidant. Its mechanism of action is currently unknown (56). It has been tested in animal models of epilepsy and is effective in the 6 Hz and maximal electroshock model (57). CBD exerted anticonvulsant effects against PTZ-induced acute generalized seizures, pilocarpine-induced temporal lobe convulsions, and penicillin- induced partial seizures in Wistar-Kyoto rats (58,59).
Cannabidivarin, a propyl analog of cannabidiol, acting on the CB2 cannabinoid receptor, has been shown to be anticonvulsant in mouse and rat models of seizures, including the maximum electroshock model, and the audiogenic seizure model in mice, and pentylenetetrazole model in rats (60). These effects were seen at doses that did not impair motor function.
Epidiolex is a natural plant-based form of cannabidiol delivered in an oil-based capsule that is in development for Dravet syndrome. Cannabidiol (Epidiolex) has been designated orphan drug status for the treatment of Dravet syndrome by the FDA. It is available in several expanded access programs. At the time of this writing, GBH Pharma is conducting an open-label dose-ranging trial in children with severe epilepsy. It is likely that cannabidivarin and other phytocannabinoids will be further investigated for specific use in human epilepsy.
Carisbamate
Carisbamate (S-2-O-carbamoyl-1-o-chlorophenyl-ethanol, formerly RWJ-333369) is a novel molecule in the carbamate class that has exhibited potent and broad activity in rodent seizure models including audiogenic seizure models and seizures induced by MES (Maximal Electroshock), PTZ, BIC (bicuculline), and picrotoxin, as well as in corneal-kindled rats (6). Carisbamate also suppressed the duration of spike-and-wave discharges in the GAERS model (61) and has shown efficacy in additional rat models. Approximately 44% of the drug is protein bound, and primary routes of metabolism include O-glucuronidation and carbamate ester hydrolysis followed by oxidation of the aliphatic side chain (62). Unlike felbamate, there is low likelihood of conversion to the reactive metabolite mercapturic acid or its conjugates (63). This is important as presence of mercapturic acid has been suggested as a potential cause of some of felbamate’s serious idiosyncratic reactions. Carisbamate has a 12-hour half-life allowing for twice daily dosing and follows linear pharmacokinetics (64). Maximum concentrations (Cmax) occur 1 to 3 hours after dosing. Oral (metabolic) clearance is low. It is primarily renally excreted. Carisbamate has minimal impact on CYP450 hepatic enzymes and only slightly increases valproic acid and lamotrigine clearance (63). While carisbamate has no effect on carbamazepine pharmacokinetics, the Cmax of carisbamate is reduced by approximately 30% when administered with carbamazepine (65). Carisbamate plasma concentration is reduced to a lesser extent when administered with an oral contraceptive (63). A randomized, double-blind, placebo-controlled, dose-ranging Phase IIb study for adjunctive use in partial-onset seizures has recently been completed (66). At carisbamate doses of 300, 800, and 1600 mg/day, patients experienced a reduced seizure frequency of 24% (P = 0.001), 21% (P = 0.006), and 29% (P < 0.001), respectively, compared to a 6% reduction in the placebo group. The most common adverse events in patients were CNS related (headaches, dizziness, somnolence) and led to drug discontinuation in 6%, 12%, and 19% in each of the respective carisbamate-treated groups (vs. 8% in the placebo group). However, two other identical randomized placebo-controlled trials revealed that patients treated with carisbamate 400 mg/day had significant improvement (P < 0.01) in percent reduction of seizure frequency compared to placebo in one study, but not the other (67). Carisbamate 200 mg/day did not differ statistically from placebo in either study (67).
Johnson and Johnson filed a new drug application (NDA) to the FDA in 2008. After initial approval, the conflicting results from the completed studies led the FDA to not give marketing approval in 2009, and development was suspended. This compound has recently been acquired by SK Life Science; it is presently unclear if it will continue in development for epilepsy. It may be useful to explore its use further in refractory idiopathic generalized epilepsy with photosensitivity.
Everolimus
Everolimus is an exciting new advance in the therapy of tuberous sclerosis complex and opens the way for more specific biologic therapies as advances in genetics and genomics uncover more specific molecular mechanisms underlying the epilepsies. Tuberous sclerosis complex is frequently associated with treatment resistant epilepsy and is the prototypic “mTORopathy” which also includes a proportion of other disorders associated with epilepsy including focal cortical dysplasia, ganglioglioma, and hemimegancephaly (68). The mTOR pathway is critical in the control of cell growth and proliferation in the development of the cerebral cortex. Aberrant control of the mTOR pathway due to pathogenic mutations in mTOR complex 1 (mTORC1) and other genes involved in the mTOR pathway may allow for abnormal cell growth and differentiation. Everolimus is a rapamycin analog (a “rapalog”) with a superior pharmacokinetic profile to that of rapamycin. It has been shown to be effective and safe in the treatment of patients with tuberous sclerosis complex and associated subependymal giant cell astrocytomas (69). The recent EXIST-1 trial (70), involving 117 patients aged between 0 and 65 years, demonstrated a >50% reduction in the volume of subependymal giant cell astrocytomas in 35% of patients on active treatment, suggesting a clear disease-modifying effect of everolimus in tuberous sclerosis complex. Adverse effects include stomatitis and mouth ulceration and were mild and did not lead to treatment discontinuation in the EXIST-1 trial. Mean duration of active treatment was 41.9 weeks. Anecdotal reports have described the beneficial effects on seizure control in patients with refractory epilepsy associated with tuberous sclerosis complex (71); trials are now in progress or planned to test this exciting prospect more formally. If this is proven, then it may be that individuals with epilepsy associated with genetic defects involving the mTOR pathway more broadly may benefit from everolimus therapy.
Huperzine A
Huperzine A, an N-methyl-d-aspartate (NMDA) receptor and acetylcholinesterase inhibitor, is a sesquiterpene lycopodium alkaloid isolated from the Chinese club moss Huperzia serrata, traditionally used in China for swelling, fever and inflammation, blood disorders, and schizophrenia (7,72,73
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


