Chemical formula: C9H8O4
SMILES: CC(=O)OC1=CC=CC=C1C(O)=O
Pharmacodynamics
ASA is an analgesic, antipyretic, antirheumatic, and anti-inflammatory agent. ASA’s mode of action as an antiinflammatory and antirheumatic agent may be due to inhibition of synthesis and release of prostaglandins. ASA appears to produce analgesia by virtue of both a peripheral and CNS effect. Peripherally, ASA acts by inhibiting the synthesis and release of prostaglandins. Acting centrally, it would appear to produce analgesia at a hypothalamic site in the brain, although the mode of action is not known. ASA also acts on the hypothalamus to produce antipyresis; heat dissipation is increased as a result of vasodilation and increased peripheral blood flow. ASA’s antipyretic activity may also be related to inhibition of synthesis and release of prostaglandins
Mechanism of action
ASA directly and irreversibly inhibits the activity of both types of cyclooxygenase (COX-1 and COX-2) to decrease the formation of precursors of prostaglandins and thromboxanes from arachidonic acid. This makes ASA different from other NSAIDS (such as diclofenac and ibuprofen) which are reversible inhibitors. Salicylate may competitively inhibit prostaglandin formation. ASA’s antirheumatic (nonsteroidal anti-inflammatory) actions are a result of its analgesic and anti-inflammatory mechanisms; the therapeutic effects are not due to pituitary-adrenal stimulation.
Platelet-derived COX-1 generates thromboxane A2 (TXA2), a potent vasoconstrictor and platelet agonist. In contrast, endothelial cell COX-1 generates prostacyclin which possesses vasodilatory and antiplatelet actions. Platelet COX-1 is more sensitive than endothelial cell COX-1 is to this effect of ASA and this distinction explains the recommendation for using very low doses of ASA to retard platelet aggregation. Inhibiting platelet COX-1 without diminishing endothelial cell COX-1 is desirable in patients with cardiovascular disease
Inhibition of platelet COX-1 results in decreased platelet aggregation, leading to a prolonged bleeding time. Hemostatic effects return to normal roughly 36 hours after the last dose of the drug. The platelet aggregation-inhibiting effect of ASA specifically involves the compound’s ability to act as an acetyl donor to COX; the nonacetylated salicylates have no clinically significant effect on platelet aggregation. Irreversible acetylation renders COX inactive, thereby preventing the formation of the aggregating agent TXA2 in platelets. Since platelets lack the ability to synthesize new proteins, the effects persist for the life of the exposed platelets (7-10 days). ASA may also inhibit production of the platelet aggregation inhibitor, prostacyclin (prostaglandin I2), by blood vessel endothelial cells; however, inhibition prostacyclin production is not permanent as endothelial cells can produce more cyclooxygenase to replace the non-functional enzyme. ASA has little effect on thrombin-induced platelet aggregation. More recently, it has been proposed that the beneficial effect of ASA might be related to its ability to reduce circulating levels of C-reactive protein. In very high and toxic doses, ASA also exerts a direct inhibitory effect on vitamin K-dependent hemostasis. Prothrombin synthesis is impaired, resulting in hypoprothrombinemia
Absorption
Absorption is generally rapid and complete following oral administration but may vary according to specific salicylate used, dosage form, and other factors such as tablet dissolution rate and gastric or intraluminal pH
Volume of distribution
Not available
Protein binding
High (99.5%) to albumin. Decreases as plasma salicylate concentration increases, with reduced plasma albumin concentration or renal dysfunction, and during pregnancy
Metabolism
ASA is rapidly hydrolyzed primarily in the liver to salicylic acid, which is conjugated with glycine (forming salicyluric acid) and glucuronic acid and excreted largely in the urine
Route of elimination
Not available
Half life
The plasma half-life is approximately 15 minutes; that for salicylate lengthens as the dose increases: doses of 300 to 650 mg have a half-life of 3.1 to 3.2 hours; with doses of 1 gram, the half-life is increased to 5 hours and with 2 grams it is increased to about 9 hours.
Clearance
Not available
Toxicity
Oral, mouse: LD50=250 mg/kg; Oral, rabbit: LD50=1010 mg/kg; Oral, rat: LD50=200 mg/kg. Effects of overdose include: tinnitus, abdominal pain, hypokalemia, hypoglycemia, pyrexia, hyperventilation, dysrhythmia, hypotension, hallucination, renal failure, confusion, seizure, coma, and death.
Drug Interactions
Drug
Interaction
Acenocoumarol
Acetylsalicylic acid increases the effect of the anticoagulant, acenocoumarol
Acetazolamide
Acetylsalicylic acid at high dose increases the effect of the carbonic anhydrase inhibitor, acetazolamide
Acetohexamide
Acetylsalicylic acid increases the effect of sulfonylurea, acetohexamide
Anisindione
Acetylsalicylic acid increases effect of the anticoagulant, anisindione
Betamethasone
The corticosteroid, betamethasone, may decrease the effect of the salicylate, acetylsalicylic acid
Chlorpropamide
Acetylsalicylic acid may increase the effect of the sulfonylurea, chlorpropamide
Cortisone acetate
The corticosteroid, cortisone acetate, may decrease the effect of the salicylate, acetylsalicylic acid
Dexamethasone
The corticosteroid, dexamethasone, may decrease the effect of the salicylate, acetylsalicylic acid
Dichlorphenamide
Acetylsalicylic acid at high dose increases the effect of the carbonic anhydrase inhibitor, dichlorphenamide
Dicumarol
Acetylsalicylic acid increases effect of the anticoagulant, dicumarol
Fludrocortisone
The corticosteroid, fludrocortisone, may decrease the effect of the salicylate, acetylsalicylic acid
Ginkgo biloba
Additive anticoagulant/antiplatelet effects may increase bleed risk. Concomitant therapy should be avoided
Gliclazide
Acetylsalicylic acid increases the effect of the sulfonylurea, gliclazide
Glipizide
Acetylsalicylic acid increases the effect of the sulfonylurea, glipizide
Glisoxepide
Acetylsalicylic acid increases the effect of the sulfonylurea, glisoxepide
Glyburide
Acetylsalicylic acid increases the effect of the sulfonylurea, glibenclamide
Glycodiazine
Acetylsalicylic acid increases the effect of sulfonylurea, glycodiazine
Griseofulvin
Griseofulvin may decrease the efficacy of acetylsalicylic acid
Heparin
Increased risk of bleeding
Hydrocortisone
The corticosteroid, hydrocortisone, may decrease the effect of the salicylate, acetylsalicylic acid
Ibuprofen
Concomitant therapy of the NSAID, ketoprofen, and acetylsalicylic acid may result in additive adverse/toxic effects (e.g. GI bleeding). The NSAID may also limit the cardioprotective effect of acetylsalicylic acid. Occasional concomitant use may not cause clinically significant problems, but regular, frequent concomitant therapy is not recommended
Ketoprofen
Concomitant therapy of the NSAID, ketoprofen, and acetylsalicylic acid may result in additive adverse/toxic effects (e.g. GI bleeding). The NSAID may also limit the cardioprotective effect of acetylsalicylic acid. Occasional concomitant use may not cause clinically significant problems, but regular, frequent concomitant therapy is not recommended
Ketorolac
Acetylsalicylic acid may increase the adverse GI effects ketorolac
Methazolamide
Acetylsalicylic acid at high dose increases the effect of the carbonic anhydrase inhibitor, methazolamide
Methotrexate
Acetylsalicylic acid increases the effect and toxicity of methotrexate
Methylprednisolone
The corticosteroid, methylprednisolone, may decrease the effect of the salicylate, acetylsalicylic acid
Paramethasone
The corticosteroid, paramethasone, may decrease the effect of the salicylate, acetylsalicylic acid
Prednisolone
The corticosteroid, prednisolone, may decrease the effect of the salicylate, acetylsalicylic acid
Prednisone
The corticosteroid, prednisone, may decrease the effect of the salicylate, acetylsalicylic acid
Probenecid
Acetylsalicylic acid decreases the uricosuric effect of probenecid
Sulindac
Risk of additive toxicity (e.g. bleed risk). Acetylsalicylic acid may decrease the serum concentration of sulindac. Sulindac may counteract the cardioprotective effects of acetylsalicylic acid. Consider alternate therapy or monitor for changes in the therapeutic and adverse effects of both agents if the interacting agent is initiated, discontinued or dose changed
Telmisartan
Concomitant use of relmisartan and acetylsalicylic acid may increase the risk of acute renal failure and hyperkalemia. Monitor renal function at the beginning and during treatment
Tiaprofenic acid
Increased risk of gastrointestinal bleeding
Ticlopidine
Increased effect of ticlopidine
Tolazamide
Acetylsalicylic acid increases the effect of the sulfonylurea, tolazamide
Tolbutamide
Acetylsalicylic acid increases the effect of the sulfonylurea, tolbutamide
Tolmetin
Additive adverse effects increase the risk of gastrointestinal bleeding. Possible decrease in the cardioprotective effect of acetylsalicylic acid. Monitor for increased bleeding risk during concomitant therapy
Trandolapril
Acetylsalicylic acid may reduce the efficacy of Trandolapril. Monitor for changes in Trandolapril efficacy if acetylsalicylic acid is initiated, discontinued or dose changed
Treprostinil
The prostacyclin analogue, Treprostinil, increases the risk of bleeding when combined with the antiplatelet agent, Acetylsalicylic acid. Monitor for increased bleeding during concomitant thearpy
Triamcinolone
The corticosteroid, triamcinolone, may decrease the effect of the salicylate, acetylsalicylic acid
Valproic acid
Acetylsalicylic acid increases the effect of valproic acid
Warfarin
The antiplatelet effects of acetylsalicylic acid may increase the bleed risk associated with warfarin
Food interactions
- Avoid alcohol, alcohol appears to cause a 50 to 100% increases in ASA serum levels
- Take with a full glass of water
- Take with food to reduce gastric irritation
Table 64.2. Aspirin (ASA) overview. SMILES indicates simplified molecular-input line-entry system [25].
Effectiveness of ASA
ASA reduces the risk of recurrent stroke and other major vascular events of approximately 13-22% (mean 13%; 95% CI 6-19%) compared with control, when administered acutely and over the long-term (Figure 64.1) [26-36]. Antiplatelet therapy reduced the risk of secondary stroke, MI, or vascular death by 22% among the subset of patients with prior cerebrovascular disease (TIA or stroke) in the 2002 ATC; the absolute benefit was 36 events prevented per 1000 patients treated for 29 months [19]. The benefit was independent of age (greater or less than 65), sex, diabetes, or hypertension. These results were further confirmed in an implemented 2009 ATC meta-analysis of 16 placebo-controlled secondary prevention trials. ASA reduced the risk of any serious vascular event and the risk of ischemic stroke of 19% and 22%, respectively [37]. In high-risk patients, stopping antiplatelet therapy may increase the risk of stroke. In one study of patients hospitalized with cerebral infarction 4.5% patients (n=13 of 289) had recently stopped antiplatelet therapy. Most had been taking ASA and in all patients, the antiplatelet agent had been discontinued within 6 to 10 days of stroke onset, consistent with the known lifespan (about 10 days) of inhibited platelets [38]. Similar results were found in a case-control study comparing 309 patients with stroke to 309 matched controls. ASA discontinuation was associated with a significantly increased risk of a recurrent cerebrovascular event (TIA or ischemic stroke; OR 3.4, 95% CI 1.08-10.63) [39]. In addition, ASA decreases the risk of other cardiovascular events in a wide range of patients with established cardiovascular disease and the risk of death from certain cancers in the long-term use. However, ASA shows a limited efficacy and treatment failures should be expected in most patients with TIA and stroke. The modest treatment effect of ASA in many patients may be attributable to non-compliance [40], and in some patients to suboptimum platelet inhibition by ASA, as shown by incomplete inhibition of TXA2 production or of platelet activation and aggregation [41,42].
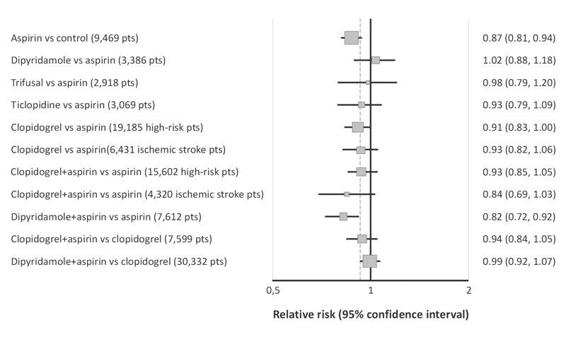
Figure 64.1. Summary of the results of RCTs of antiplatelet drugs in patients with TIA or ischemic stroke of presumed arterial origin. Relative effects of antiplatelet regimens vs.placebo, aspirin, and clopidogrel in reducing the risk of stroke, myocardial infarction, or vascular death (major vascular events). The x-axis shows the degree of relative risk of stroke, myocardial infarction, or vascular events/death with each antiplatelet regimen. Point estimates and 95% CIs are shown. The data derived from a systematic review of all trials [26-31,33,35,36].
Dose of ASA
The lack of proof of an optimal dose is a controversial issue concerning the efficacy of ASA in stroke prevention. In secondary stroke prevention studies, ASA doses range between 20 to 1300 mg; however, 50 to 325 mg/day of ASA is as effective as higher dose in most studies [11,43-48]. Lower doses within this range appear to provide the same benefit as higher doses [43-48]. Doses of 75 to 150 mg/day produced the same risk reduction, compared with placebo, as doses of 150 to 325 mg/day according to a review of 195 trials of secondary prevention by the ATC [11]. The ATC analysis of trials directly comparing ASA<75 mg/day to ASA ≥75 mg/day, showed comparable effectiveness between the two regimens. However, the ASA doses of<75 mg/day have been less widely assessed than doses of 75 to 150 mg/day, so uncertainty remains regarding the effectiveness of doses<75 mg/day compared with higher ASA doses in the ATC meta-analysis. No additional benefit with a greater risk of non-fatal major gastrointestinal hemorrhage was demonstrated in two further trials comparing higher with lower doses of ASA regimens [45,49]. The risk of major stroke, MI, or vascular death was 15% less with ASA than with placebo in the United Kingdom Transient Ischemic Attack trial (UK-TIA); in this trial, patients with minor ischemic stroke or TIA were randomized to 600 mg ASA twice daily, 300 mg ASA once daily, or placebo. The two ASA doses were equal in efficacy. Also in the Dutch TIA trial of patients with TIA or non-disabling stroke, there was no difference in stroke prevention between the two doses of ASA 30 mg ASA vs. 283 mg in preventing vascular death, non-fatal stroke, or non-fatal MI. Again, however, the 30 mg dose group had fewer bleeding complications [45]. No clinical trial evidence supports an increase in the ASA dose for patients who have a stroke while receiving ASA. Lower doses may also be effective. The Dutch TIA Trial [49] demonstrated a similar efficacy for stroke prevention with 30 mg compared with 283 mg of ASA per day in patients who had had a TIA or minor ischemic stroke. Similarly, in the European Stroke Prevention Study-2 (ESPS-2), 50 mg of ASA daily reduced stroke risk by 18% compared with placebo (29 strokes prevented per 1000 treated), an effect of comparable magnitude to the other trials cited above [47]. This benefit is consistent with laboratory observations that 30 mg of ASA per day results in complete suppression of TXA2 production [50]. Both the Food and Drug Administration and the American College of Cardiology now support ASA doses between 50 and 325 mg, either alone or in combination with extended-release DP (ER-DP) for prevention of recurrent noncardioembolic TIA or stroke [51,52]. ASA in doses as low as 75 mg/day reduces risk of vascular events in patients with previous stroke or TIA [17,18].
Toxicity and Risk of Bleeding
Low-dose ASA are associated with low risk of major bleeding (number needed to harm [NNH] 769 per year) [20]. In the UK TIA trial, for example, gastrointestinal hemorrhage occurred in 1.6% of patients on placebo, 2.6% on 300 mg ASA, and 4.7% on 1200 mg ASA [43]. ASA doses ≤200 mg/day were associated with a significantly lower rate of major bleeding events compared with higher doses in an analysis of data from 31 randomized, controlled trials [53]. However, major bleedings were similar when ASA <100 mg/day was compared with 100 to 200 mg/day, the overall rate of bleeding complications (including major, minor and insignificant events) was associated with a lower risk in ASA <100 mg/day group compared with the 100 to 200 mg/day and >200 mg/day groups. A later meta-analysis of 22 randomized trials of low-dose ASA (75 to 325 mg/day) vs.placebo for cardiovascular prophylaxis reached similar conclusions within the low-dose range [20]. Compared with placebo, ASA increased the relative risk of any major bleeding, major gastrointestinal bleeding, and intracranial bleeding by 1.7- to 2.1-fold. However, the absolute annual increase in risk for any major bleeding episode (mostly gastrointestinal) and for intracranial bleeding was 0.13 and 0.03%, respectively. Furthermore, there was no evidence of an increased risk of bleeding with “high” low-dose ASA (>162 to 325 mg/day) compared with “low” low-dose ASA (75 to 162 mg/day). Although ASA increases the risk of major bleeding by about 70% (risk ratio [RR] 1.71 [95% CI 1.41-2.08]), the absolute annual increase is modest (0.13% [0.08-0.20%]), indicating that one additional major bleeding episode will occur each year for every 769 patients (95% CI 500-1250) treated with ASA [20]. The increased risk of bleeding is mainly due to an increase in major gastrointestinal bleeding (RR 2.07 [95% CI 1.61-2.66]; absolute annual increase 0.12% [0.07-0.19]) and intracranial bleeding (RR 1.65 [1.06-5.99]; absolute annual increase 0.03% [0.01-0.08]) [20]. Gastrointestinal hemorrhage was more common with the 1200 mg dose than the 300 mg dose. ASA increases risk of hemorrhagic stroke, but overall benefit on MI and ischemic stroke incidence may outweigh its adverse effects on risk of hemorrhagic stroke in most populations. ASA use associated with absolute risk increase in hemorrhagic stroke of 12 events per 10,000 persons (NNH 833) [21].
Nonresponse/resistance
About 10% of general population is resistant to antiplatelet effects of ASA [54]. Decreasing response to ASA is correlated independently with increasing risk of cardiovascular events. In a systematic review of 13 prospective cohort studies and 3 case-control studies among patients treated with ASA for secondary prevention of cardiovascular events, 5% to 65% range of ASA resistance by ex vivo nonresponsiveness to ASA based on any test of platelet TXA2 synthesis or platelet function, in a meta-analysis of 12 studies with 1813 patients 30.4% patients with vs. 12.7% patients without ASA resistance had cardiovascular events (p<0.001) although this meta-analysis is limited by heterogeneity (p=0.03) [55]. Studies showing that ASA is unable to protect against the onset of thrombotic complications. A high percentage (up to a barely believable 45%, depending on the study) of patients presents “treatment failure” or “ASA resistance,” being more common in elderly people and women [41]. The mechanisms involved in ASA resistance are probably multifactorial and include the factors shown in Figure 64.2.
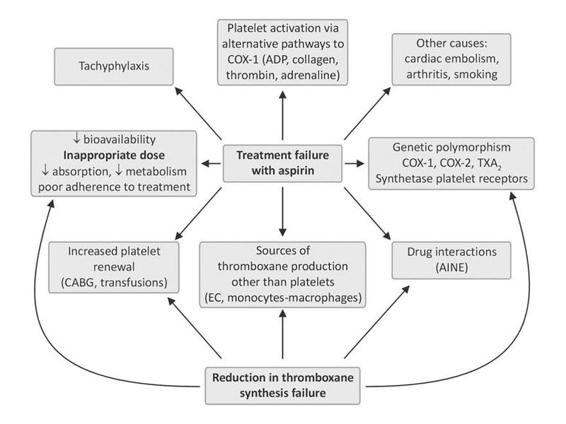
Figure 64.2. Possible mechanisms involved in aspirin resistance. NSAIDS indicates non-steroid antiinflammatories; CABG, coronary artery bypass.
Potential causes of a reduced response to ASA include inadequate dose, genetic polymorphisms of PTGS1 (which encodes prostaglandin G/H synthase 1, formerly cyclo-oxygenase 1) and other genes involved in TX biosynthesis, upregulation of non-platelet sources of TX biosynthesis, increased platelet turnover, and drug interactions (eg, with non-steroidal anti-inflammatory drugs [NSAIDs]), and occasional ASA allergy. Concomitant NSAIDs may be associated with ASA resistance. Concomitant NSAID use is associated with increased risk of ASA nonresponsiveness as determined by arachidonate acid agonist (OR 2.17, p=0.036) and by determined by collagen agonist (OR 2.67, p=0.001) [23,41,56]. The specific NSAIDs that may lead to a less than expected inhibition of platelet function by ASA are ibuprofen, naproxen, and possibly other non-selective NSAIDs, because they compete with ASA for access to its target (the active site at position 530 of a serine residue in the core of the cyclo-oxygenase enzyme) [57]. ASA hyporesponsiveness can potentially be overcome by increasing the dose or frequency of administration, and by avoiding drugs that interact with ASA. However, the benefit of these measures on clinical outcomes remains unproven. ASA resistance represents a controversial topic also because there is poor agreement on the best measurement of such resistance, as the methods of assaying platelet inhibition do not always agree, and there has also been little control for lack of adherence to the ASA regimen [40]. Likewise, switching to alternative antiplatelet agents has not been studied for ASA failures. In clinical practice ASACheck® and ASAWorks® are tests for ASA resistance which measure urine 11-dehydro-TXB2, high levels might suggest ASA resistance but could be due to other sources of TX production. Ultegra RPFA-ASA® is another point-of-care optical platelet function assay for ASA resistance. Laboratory ASA resistance is associated with increased risk of recurrent cardiovascular events in patients taking ASA for secondary prevention. To date, small studies have been conducted showing that incomplete TX synthesis suppression, despite sufficient doses (aspirin resistance), is a potential cardiovascular risk marker [58]. In the WARSS trial [59], a clinical trial of ASA vs. warfarin in non-cardioembolic stroke patients, the two agents had nonsignificant difference in efficacy. Moreover, patients who had already failed ASA when they had their index strokes had a higher risk of recurrent stroke in the course of the trial, but there was still no evidence of superior efficacy of warfarin over ASA. Established alternatives to ASA include clopidogrel and the combination of ASA and ER-DP.
Recommendations for ASA
American College of Chest Physicians (ACCP) (Ninth Edition) [15]
- Following noncardioembolic stroke or transient ischemic attack (TIA)
- ASA 75-100 mg once daily recommended as an option for long-term antiplatelet therapy over: no antiplatelet therapy (ACCP Grade 1A), oral anticoagulants (ACCP Grade 1B), combination of clopidogrel plus ASA (ACCP Grade 1B), Triflusal (ACCP Grade 2B).
- Among the recommended antiplatelet regimens, clopidogrel or combination of ASA plus extended-release DP suggested over ASA alone (ACCP Grade 2B) or triflusal (ACCP Grade 2C).
- Benefit of clopidogrel over ASA for preventing major vascular events may be offset with long-term use (>5 years) by lower cancer-related mortality with ASA.
- ASA 75-100 mg once daily recommended as an option for long-term antiplatelet therapy over: no antiplatelet therapy (ACCP Grade 1A), oral anticoagulants (ACCP Grade 1B), combination of clopidogrel plus ASA (ACCP Grade 1B), Triflusal (ACCP Grade 2B).
- Following cardioembolic stroke or TIA (that is, atrial fibrillation, including paroxysmal atrial fibrillation):
- OAT recommended over: no antithrombotic therapy (ACCP Grade 1A), ASA (ACCP Grade 1B), combination therapy with ASA plus clopidogrel (ACCP Grade 1B).
- For patients unsuitable for or who choose not to take an oral anticoagulant (for reasons other than concerns about major bleeding), combination therapy with ASA plus clopidogrel recommended over ASA (ACCP Grade 1B).
- Use of ASA as bridging therapy suggested until anticoagulation reaches therapeutic level.
- OAT recommended over: no antithrombotic therapy (ACCP Grade 1A), ASA (ACCP Grade 1B), combination therapy with ASA plus clopidogrel (ACCP Grade 1B).
American Heart Association (AHA) [13]:
- For patients with noncardioembolic ischemic stroke or TIA, antiplatelet agents recommended rather than OAT (AHA/ASA Class I, Level A):
- Acceptable options include: ASA 50-325 mg daily (AHA/ASA Class I, Level A), combination ASA 25 mg twice daily and extended-release DP 200 mg twice daily (AHA/ASA Class I, Level B), clopidogrel 75 mg daily (AHA/ASA Class iia, Level B).
- Selection of antiplatelet agent should be individualized based on patient risk factors, cost, tolerance and other clinical factors.
- Addition of ASA to clopidogrel increases risk of hemorrhage and is not routinely recommended for secondary prevention of stroke (AHA/ASA Class III, Level A).
- Clopidogrel reasonable for patients allergic to ASA (AHA/ASA Class iia, Level C).
- For patients who have ischemic stroke while taking ASA (AHA/ASA Class iib, Level C): no evidence for additional efficacy with increasing dose of ASA, alternative antiplatelet agents often considered, but no regimen specifically studied.
- Acceptable options include: ASA 50-325 mg daily (AHA/ASA Class I, Level A), combination ASA 25 mg twice daily and extended-release DP 200 mg twice daily (AHA/ASA Class I, Level B), clopidogrel 75 mg daily (AHA/ASA Class iia, Level B).
- For patients with ischemic stroke or TIA and paroxysmal or permanent atrial fibrillation:
- If unable to take oral anticoagulants, ASA alone is recommended (AHA/ASA Class I, Level A).
- Clopidogrel with ASA not recommended for patients with hemorrhagic contraindication to warfarin (AHA/ASA Class III, Level B).
- If unable to take oral anticoagulants, ASA alone is recommended (AHA/ASA Class I, Level A).
- Optimal medical therapy for patients with extracranial arterial disease (carotid or vertebrobasilar) and TIA or stroke should include antiplatelet therapy (AHA/ASA Class I, Level B).
- For patients with stroke or TIA due to 50%-99% stenosis of major intracranial artery, ASA 50-325 mg/day is recommended over warfarin (AHA/ASA Class I, Level B).
- For patients with cardiomyopathy:
- Insufficient evidence for use of warfarin in patients with prior stroke or TIA, sinus rhythm, cardiomyopathy and ejection fraction ≤ 35% (AHA/ASA Class iib, Level B).
- Treatments to consider for patients with ischemic stroke or TIA and cardiomyopathy include (AHA/ASA Class IIb, Level B): warfarin (INR 2-3), ASA 81 mg/day, clopidogrel 75 mg/day, combination of ASA 25 mg twice daily and extended-release DP 200 mg twice daily.
- Insufficient evidence for use of warfarin in patients with prior stroke or TIA, sinus rhythm, cardiomyopathy and ejection fraction ≤ 35% (AHA/ASA Class iib, Level B).
- For patients with native valvular heart disease:
- Antiplatelet therapy may be considered for patients with ischemic stroke or TIA and: native aortic or nonrheumatic mitral valve disease and no atrial fibrillation (AHA/ASA Class IIb, Level C), mitral annular calcification (AHA/ASA Class IIb, Level C), mitral valve prolapse (AHA/ASA Class IIb, Level C).
- To avoid additional bleeding risk, antiplatelet agents should not be routinely added to warfarin (AHA/ASA Class III, Level C).
- Antiplatelet therapy may be considered for patients with ischemic stroke or TIA and: native aortic or nonrheumatic mitral valve disease and no atrial fibrillation (AHA/ASA Class IIb, Level C), mitral annular calcification (AHA/ASA Class IIb, Level C), mitral valve prolapse (AHA/ASA Class IIb, Level C).
- For patients with ischemic stroke or TIA due to extracranial arterial dissection (carotid or vertebral):
- Antithrombotic therapy for at least 3-6 months is reasonable (AHA/ASA Class IIa, Level B).
- Relative efficacy of antiplatelet therapy vs. Anticoagulation is unknown (AHA/ASA Class iib, Level B).
- Antithrombotic therapy for at least 3-6 months is reasonable (AHA/ASA Class IIa, Level B).
- For patients with patent foramen ovale (PFO):
- Antiplatelet therapy is reasonable for patients with ischemic stroke or TIA and PFO (AHA/ASA Class IIa, Level B).
- Insufficient data to determine whether anticoagulation is as or more effective than ASA for secondary stroke prevention in patients with PFO (AHA/ASA Class IIb, Level B).
- Antiplatelet therapy is reasonable for patients with ischemic stroke or TIA and PFO (AHA/ASA Class IIa, Level B).
Canadian Stroke Network (CSN) [16]:
- All patients with ischemic stroke should be prescribed antiplatelet therapy for secondary prevention of recurrent stroke unless indication for anticoagulation (CSN Grade A).
- Medications include (CSN Grade A):
- ASA 80-325 mg/day (1-5 mg/kg/day in children [CSN Grade B]).
- Combined ASA (25 mg) and extended-release DP (200 mg) twice daily.
- Clopidogrel 75 mg/day.
- ASA 80-325 mg/day (1-5 mg/kg/day in children [CSN Grade B]).
- Clopidogrel may be considered an alternative in pediatric patients with contraindication to ASA (CSN Grade C).
- Long term combination of ASA and clopidogrel is not recommended for secondary stroke prevention unless there is a compelling indication (CSN Grade B).
Thienopyridine Derivatives
Ticlopidine and clopidogrel decrease platelet aggregation by inhibiting the binding of adenosine 5’-diphosphate (ADP) receptor antagonists that inhibit ADP-induced fibrinogen binding to platelets [60]. Significant inhibition is present after 2 to 3 days of therapy with ticlopidine 500 mg/day or clopidogrel 75 mg/day, with maximal inhibition occurring between 4 and 7 days. As with ASA, the antiplatelet action persists for 7 to 10 days after therapy is stopped lasting for the lifespan of the platelet.
Ticlopidine
Ticlopidine is a thienopyridine structurally and pharmacologically similar to clopidogrel (Table 64.3) [25]. The active metabolite of ticlopidine prevents binding of adenosine diphosphate (ADP) to its platelet receptor, impairing the ADP-mediated activation of the glycoprotein GPIIb/IIIa complex. It is proposed that the inhibition involves a defect in the mobilization from the storage sites of the platelet granules to the outer membrane. No direct interference occurs with the GPIIb/IIIa receptor. As the glycoprotein GPIIb/IIIa complex is the major receptor for fibrinogen, its impaired activation prevents fibrinogen binding to platelets and inhibits platelet aggregation. By blocking the amplification of platelet activation by released ADP, platelet aggregation induced by agonists other than ADP is also inhibited by the active metabolite of ticlopidine.
Structure |
Chemical formula: C14H14ClNS; SMILES: ClC1=CC=CC=C1CN1CCC2=C(C1)C=CS2 | |
Mechanism of action | The active metabolite of ticlopidine prevents binding of adenosine diphosphate (ADP) to its platelet receptor, impairing the ADP-mediated activation of the glycoprotein GPIIb/IIIa complex. It is proposed that the inhibition involves a defect in the mobilization from the storage sites of the platelet granules to the outer membrane. No direct interference occurs with the GPIIb/IIIa receptor. As the glycoprotein GPIIb/IIIa complex is the major receptor for fibrinogen, its impaired activation prevents fibrinogen binding to platelets and inhibits platelet aggregation. By blocking the amplification of platelet activation by released ADP, platelet aggregation induced by agonists other than ADP is also inhibited by the active metabolite of ticlopidine | |
Absorption | Absorption is greater than 80%. Food increases absorption | |
Volume of distribution | Not available | |
Protein binding | Binds reversibly (98%) to plasma proteins, mainly to serum albumin and lipoproteins. The binding to albumin and lipoproteins is nonsaturable over a wide concentration range. Ticlopidine also binds to alpha-1 acid glycoprotein. At concentrations attained with the recommended dose, only 15% or less ticlopidine in plasma is bound to this protein | |
Metabolism | Metabolized extensively by the liver; only trace amounts of intact drug are detected in the urine. At least 20 metabolites have been identified. It has been proposed that 1 or more active metabolites may account for ticlopidine’s activity, because ticlopidine itself is an extremely weak platelet aggregation inhibitor in vitro at the concentrations achieved in vivo. However, no active metabolite has been identified | |
Route of elimination | Ticlopidine hydrochloride is metabolized extensively by the liver; only trace amounts of intact drug are detected in the urine. Approximately 1/3 of the dose excreted in the feces is intact ticlopidine hydrochloride, possibly excreted in the bile | |
Half life | Half-life following a single 250-mg dose is approximately 7.9 hours in subjects 20 to 43 years of age and 12.6 hours in subjects 65 to 76 years of age. With repeated dosing (250 mg twice a day), half-life is about 4 days in subjects 20 to 43 years of age and about 5 days in subjects 65 to 76 years of age | |
Clearance | Not available | |
Toxicity | Single oral doses of ticlopidine at 1600 mg/kg and 500 mg/kg were lethal to rats and mice, respectively. Symptoms of acute toxicity were GI hemorrhage, convulsions, hypothermia, dyspnea, loss of equilibrium and abnormal gait | |
Drug Interactions | Drug | Interaction |
Acetylsalicylic acid | Increased effect of ticlopidine | |
Alteplase | Increased bleeding risk. Monitor for signs of bleeding | |
Ambrisentan | Ticlopidine may decrease the metabolism and clearance of ambrisentan. Consider alternate therapy or monitor for adverse/toxic effects of ambrisentan if ticlopidine is initiated, discontinued or dose changed. | |
Aminophylline | Ticlopidine increases the effect and toxicity of theophylline | |
Bortezomib | Ticlopidine may decrease the metabolism and clearance of bortezomib. Consider alternate therapy or monitor for adverse/toxic effects of bortezomib if ticlopidine is initiated, discontinued or dose changed | |
Carbamazepine | Ticlopidine increases the effect of carbamazepine | |
Carisoprodol | Ticlopidine may decrease the metabolism and clearance of carisoprodol. Consider alternate therapy or monitor for adverse/toxic effects of carisoprodol if ticlopidine is initiated, discontinued or dose changed | |
Cilostazol | Ticlopidine may decrease the metabolism and clearance of cilostazol. Consider alternate therapy or monitor for adverse/toxic effects of cilostazol if ticlopidine is initiated, discontinued or dose changed | |
Cimetidine | Cimetidine may increase ticlopidine levels. Avoid concomitant therapy. | |
Citalopram | Ticlopidine may decrease the metabolism and clearance of citalopram. Consider alternate therapy or monitor for adverse/toxic effects of citalopram if ticlopidine is initiated, discontinued or dose changed | |
Clobazam | Ticlopidine may decrease the metabolism and clearance of clobazam. Consider alternate therapy or monitor for adverse/toxic effects of clobazam if ticlopidine is initiated, discontinued or dose changed | |
Clomipramine | Ticlopidine may decrease the metabolism and clearance of clomipramine. Consider alternate therapy or monitor for adverse/toxic effects of clomipramine if ticlopidine is initiated, discontinued or dose changed | |
Cyclosporine | Ticlopidine decreases the effect of cyclosporine | |
Diazepam | Ticlopidine may decrease the metabolism and clearance of diazepam. Consider alternate therapy or monitor for adverse/toxic effects of diazepam if ticlopidine is initiated, discontinued or dose changed | |
Digoxin | Ticlopidine may decrease digoxin levels. Monitor for digoxin levels with ticlopidine is initiated, discontinued or dose changed | |
Dyphylline | Ticlopidine increases the effect and toxicity of theophylline | |
Escitalopram | Ticlopidine may decrease the metabolism and clearance of escitalopram. Consider alternate therapy or monitor for adverse/toxic effects of ambrisentan if escitalopram is initiated, discontinued or dose changed | |
Ethotoin | Ticlopidine increases the effect of hydantoin | |
Fosphenytoin | Ticlopidine may decrease the metabolism and clearance of fosphenytoin. Consider alternate therapy or monitor for adverse/toxic effects of fosphenytoin if ticlopidine is initiated, discontinued or dose changed | |
Ginkgo biloba | Additive anticoagulant/antiplatelet effects may increase bleed risk. Concomitant therapy should be avoided | |
Heparin | Increased bleeding risk. Monitor aPTT | |
Ifosfamide | Ticlopidine may decrease the metabolism and clearance of ifosfamide. Consider alternate therapy or monitor for adverse/toxic effects of ifosfamide if ticlopidine is initiated, discontinued or dose changed | |
Imipramine | Ticlopidine may decrease the metabolism and clearance of Imipramine. Consider alternate therapy or monitor for adverse/toxic effects of Imipramine if ticlopidine is initiated, discontinued or dose changed | |
Mephenytoin | Ticlopidine increases the effect of hydantoin | |
Methsuximide | Ticlopidine may decrease the metabolism and clearance of methsuximide. Consider alternate therapy or monitor for adverse/toxic effects of methsuximide if ticlopidine is initiated, discontinued or dose changed | |
Moclobemide | Ticlopidine may decrease the metabolism and clearance of moclobemide. Consider alternate therapy or monitor for adverse/toxic effects of moclobemide if ticlopidine is initiated, discontinued or dose changed | |
Nilutamide | Ticlopidine may decrease the metabolism and clearance of nilutamide. Consider alternate therapy or monitor for adverse/toxic effects of nilutamide if ticlopidine is initiated, discontinued or dose changed | |
Oxtriphylline | Ticlopidine increases the effect and toxicity of theophylline | |
Pentamidine | Ticlopidine may decrease the metabolism and clearance of pentamidine. Consider alternate therapy or monitor for adverse/toxic effects of pentamidine if ticlopidine is initiated, discontinued or dose changed | |
Phenobarbital | Ticlopidine may decrease the metabolism and clearance of phenobarbital. Consider alternate therapy or monitor for adverse/toxic effects of phenobarbital if ticlopidine is initiated, discontinued or dose changed | |
Phenytoin | Ticlopidine may decrease the metabolism and clearance of phenytoin. Consider alternate therapy or monitor for adverse/toxic effects of phenytoin if ticlopidine is initiated, discontinued or dose changed | |
Reteplase | Increased bleeding risk. Monitor for signs of bleeding | |
Sodium bicarbonate | Sodium bicarbonate may decrease ticlopidine levels. Administer agents 1 to 2 hours apart | |
Streptokinase | Increased bleeding risk. Monitor for signs of bleeding | |
Tamoxifen | Ticlopidine may decrease the therapeutic effect of tamoxifen by decreasing the production of active metabolites. Consider alternate therapy | |
Tamsulosin | Ticlopidine, a CYP2D6 inhibitor, may decrease the metabolism and clearance of Tamsulosin, a CYP2D6 substrate. Monitor for changes in therapeutic/adverse effects of tamsulosin if ticlopidine is initiated, discontinued, or dose changed | |
Tenecteplase | Increased bleeding risk. Monitor for signs of bleeding | |
Theophylline | Ticlopidine increases the effect and toxicity of theophylline | |
Thioridazine | Ticlopidine may decrease the metabolism of thioridazine. Concomitant therapy is contraindicated | |
Tizanidine | Ticlopidine may decrease the metabolism and clearance of tizanidine. Consider alternate therapy or use caution during co-administration | |
Tramadol | Ticlopidine may decrease the effect of tramadol by decreasing active metabolite production | |
Treprostinil | The prostacyclin analogue, Treprostinil, increases the risk of bleeding when combined with the antiplatelet agent, Ticlopidine. Monitor for increased bleeding during concomitant thearpy | |
Trimipramine | The strong CYP2C19 inhibitor, ticlopidine, may decrease the metabolism and clearance of trimipramine, a CYP2C19 substrate. Consider alternate therapy or monitor for changes in therapeutic and adverse effects of trimipramine if ticlopidine is initiated, discontinued or dose changed | |
Warfarin | Increased bleeding risk. Monitor INR | |
Class effects | Thienopyridines may be slightly more effective than ASA for preventing serious vascular events in high-risk patients (Level 2 [mid-level] evidence) [35] | |
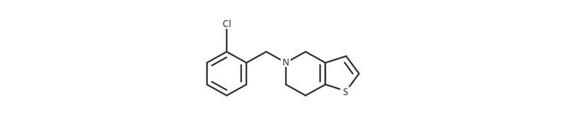
Table 64.3. Ticlopidine overview. SMILES indicates simplified molecular-input line-entry system.
When taken orally, ticlopidine causes a time- and dose-dependent inhibition of both platelet aggregation and release of platelet granule constituents, as well as, a prolongation of bleeding time. The intact drug has nonsignificant in vitro activity at the concentrations attained in vivo; and, although analysis of urine and plasma indicates at least 20 metabolites, no metabolite which accounts for the activity of ticlopidine has been isolated. Ticlopidine was initially approved for the secondary prevention of stroke and was subsequently found to be beneficial for other types of vascular events [61]. It is typically administered twice daily with food. It is associated with a significant risk for severe neutropenia (approximately 1%), mainly in the first 2-3 months of treatment. A lesser risk of thrombotic thrombocytopenic purpura is also present. Because of these hematological adverse effects, complete blood counts are recommended every 2 weeks for the first 3 months that patients are on the drug [61]. Diarrhea occurs in more than 20% of patients as well. These side effects have significantly limited its use.
Effectiveness of ticlopidine. Three major trials involving patients with TIA and stroke evaluated its role in stroke prevention. The Canadian American Ticlopidine Study, which included 1072 patients with ischemic stroke, compared ticlopidine with placebo [62]. Patients in the ticlopidine group had a significant 23.3% reduction in the combined end point of stroke, MI, or vascular death compared with the placebo group (11.3% per year vs. 14.8% per year; p=0.02). The CATS trial compared ticlopidine vs.placebo in patients who had suffered a significant stroke. At a mean of 24 months follow-up, the primary composite end point of stroke, MI, and vascular death was significantly lower with ticlopidine compared with placebo (10.8% vs. 15.3%, relative risk reduction [RRR] 30%) [62]. Analysis by intention-to-treat gave a smaller estimate of RRR for stroke, MI, or vascular death (23%). The Ticlopidine ASA Stroke Study (TASS) trial compared ticlopidine (500 mg/day) to ASA (1300 mg/day) in 3,069 patients with a recent TIA or mild stroke [63]. There was a 12% reduction in the primary end point of nonfatal stroke or death in patients receiving ticlopidine compared with those receiving ASA (17% vs. 19%; p=0.048). Ticlopidine treatment was also associated with a significant reduction in the rate of fatal and nonfatal stroke compared with ASA (10% vs. 13%, respectively; RRR 21% [95% CI 4-38]). A TASS subgroup analysis showed a larger benefit for ticlopidine in African Americans prompting the African American Antiplatelet Stroke Prevention Study (AAASPS), in which ticlopidine was compared with ASA for secondary stroke prevention in African American stroke/TIA patients [64]. The AAASPS trial compared ticlopidine (500 mg/day) to ASA (650 mg/day) in 1809 black patients with noncardioembolic ischemic stroke [64]. At two-year follow-up, there was nonsignificant difference in the primary end point (stroke, MI, vascular death) between ticlopidine and ASA in African Americans. However, for the end point of fatal or non-fatal stroke, there was a trend favoring ASA over ticlopidine. The hematologic side effects and the availability of clopidogrel have significantly reduced the use of ticlopidine for recurrent stroke prevention in patients with ischemic stroke. Despite the evidence of benefit in the CATS and TASS trials, ticlopidine is not considered a first-line antiplatelet agent for stroke prevention because of side effects and relatively high cost.
Side effects of ticlopidine. Severe neutropenia, which occurs in approximately 1% of patients is the most serious complication of ticlopidine therapy. Thus, for the first three months of treatment, patients must undergo biweekly complete blood counts. This side effect limits the utility of ticlopidine. Other common side effects, which occur more frequently with ticlopidine than ASA, are rash and diarrhea.
Clopidogrel
Clopidogrel is a thienopyridine that inhibits ADP-dependent platelet aggregation (Table 64.4) [60]. Clopidogrel, an antiplatelet agent structurally and pharmacologically similar to ticlopidine, is used to reduce atherosclerotic events such as myocardial infarction, stroke, and vascular death in patients who have had a recent stroke, recent MI, or have established peripheral vascular disease. The active metabolite of clopidogrel prevents binding of adenosine diphosphate (ADP) to its platelet receptor, impairing the ADP-mediated activation of the glycoprotein GPIIb/IIIa complex. It is proposed that the inhibition involves a defect in the mobilization from the storage sites of the platelet granules to the outer membrane. No direct interference occurs with the GPIIb/IIIa receptor. As the glycoprotein GPIIb/IIIa complex is the major receptor for fibrinogen, its impaired activation prevents fibrinogen binding to platelets and inhibits platelet aggregation. By blocking the amplification of platelet activation by released ADP, platelet aggregation induced by agonists other than ADP is also inhibited by the active metabolite of clopidogrel.
Structure |
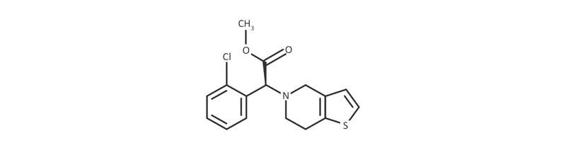
Chemical formula: C16H16ClNO2S SMILES: COC(=O)[C@@H](N1CCC2=C(C1)C=CS2)C1=CC=CC=C1Cl | |
Pharmacodynamics | Clopidogrel, an antiplatelet agent structurally and pharmacologically similar to ticlopidine, is used to reduce atherosclerotic events such as myocardial infarction, stroke, and vascular death in patients who have had a recent stroke, recent MI, or have established peripheral vascular disease | |
Mechanism of action | The active metabolite of clopidogrel prevents binding of adenosine diphosphate (ADP) to its platelet receptor, impairing the ADP-mediated activation of the glycoprotein GPIIb/IIIa complex. It is proposed that the inhibition involves a defect in the mobilization from the storage sites of the platelet granules to the outer membrane. he drug specifically and irreversibly inhibits the P2Y12 subtype of ADP receptor, which is important in aggregation of platelets and cross-linking by the protein fibrin. No direct interference occurs with the GPIIb/IIIa receptor. As the glycoprotein GPIIb/IIIa complex is the major receptor for fibrinogen, its impaired activation prevents fibrinogen binding to platelets and inhibits platelet aggregation. By blocking the amplification of platelet activation by released ADP, platelet aggregation induced by agonists other than ADP is also inhibited by the active metabolite of clopidogrel | |
Absorption | Absorption is at least 50% based on urinary excretion of clopidogrel-related metabolites. Bioavailability has not been found to be affected by food. | |
Volume of distribution | Not available | |
Protein binding | 98% | |
Metabolism | Hepatic, extensive and rapid, by hydrolysis to the main circulating metabolite, a carboxylic acid derivative, which accounts for approximately 85% of the circulating drug-related compounds. A glucuronic acid derivative of the carboxylic acid derivative has also been found in plasma and urine. Neither the parent compound nor the carboxylic acid derivative has a platelet inhibiting effect | |
Route of elimination | Following an oral dose of 14C-labeled clopidogrel in humans, approximately 50% of total radioactivity was excreted in urine and approximately 46% in feces over the 5 days post-dosing | |
Half life | Carboxylic acid derivative: 8 hours (after single and multiple doses). Covalent binding to platelets has accounted for 2% of radiolabeled clopidogrel with a half-life of 11 days | |
Clearance | Not available | |
Toxicity | Symptoms of acute toxicity include vomiting (in baboons), prostration, difficult breathing, and gastrointestinal hemorrhage | |
Drug interactions | Drug | Interaction |
Bortezomib | Moderate CYP2C19 Inhibitors like bortezomib may decrease serum concentrations of the active metabolite(s) of Clopidogrel. Avoid concurrent use of moderate CYP2C19 inhibitors with clopidogrel whenever possible. If such a combination must be used, monitor closely for evidence of reduced clinical response to clopidogrel | |
Drotrecogin alfa | Antiplatelet agents such as clopidogrel may enhance the adverse/toxic effect of Drotrecogin Alfa. Bleeding may occur. Increase monitoring for signs/symptoms of bleeding during concomitant therapy. If possible, avoid use of drotrecogin within 7 days of use of any IIb/IIIa antagonists, higher dose aspirin (more than 650 mg/day), or use of other antiplatelet agents | |
Esomeprazole | Esomeprazole may decrease serum concentrations of the active metabolite(s) of clopidogrel. Due to the possible risk for impaired clopidogrel effectiveness with this combination, clinicians should carefully consider the need for concurrent esomeprazole therapy in patients receiving clopidogrel. Monitor response to clopidogrel closely when using clopidogrel with esomeprazole. Whether there are differences among individual proton pump inhibitors is unclear. Other acid-lowering therapies (e.g., H2-receptor antagonists, antacids, etc.) do not appear to share this interaction with clopidogrel | |
Ginkgo biloba | Additive anticoagulant/antiplatelet effects may increase bleed risk. Concomitant therapy should be avoided | |
Lansoprazole | Lansoprazole may decrease serum concentrations of the active metabolite(s) of clopidogrel. Due to the possible risk for impaired clopidogrel effectiveness with this combination, clinicians should carefully consider the need for concurrent lansoprazole therapy in patients receiving clopidogrel. Monitor response to clopidogrel closely when using clopidogrel with lansoprazole. Whether there are differences among individual proton pump inhibitors is unclear. Other acid-lowering therapies (e.g., H2-receptor antagonists, antacids, etc.) do not appear to share this interaction with clopidogrel | |
Omeprazole | Omeprazole may decrease serum concentrations of the active metabolite(s) of clopidogrel. Clopidogrel prescribing information recommends avoiding concurrent use with omeprazole, due to the possibility that combined use may result in decreased clopidogrel effectiveness | |
Pantoprazole | Pantoprazole may decrease serum concentrations of the active metabolite(s) of clopidogrel. Due to the possible risk for impaired clopidogrel effectiveness with this combination, clinicians should carefully consider the need for concurrent pantoprazole therapy in patients receiving clopidogrel. Monitor response to clopidogrel closely when using clopidogrel with pantoprazole. Whether there are differences among individual proton pump inhibitors is unclear. Other acid-lowering therapies (e.g., H2-receptor antagonists, antacids, etc.) do not appear to share this interaction with clopidogrel | |
Rabeprazole | Rabeprazole may decrease serum concentrations of the active metabolite(s) of clopidogrel. Due to the possible risk for impaired clopidogrel effectiveness with this combination, clinicians should carefully consider the need for concurrent rabeprazole therapy in patients receiving clopidogrel. Monitor response to clopidogrel closely when using clopidogrel with rabeprazole. Whether there are differences among individual proton pump inhibitors is unclear. Other acid-lowering therapies (e.g., H2-receptor antagonists, antacids, etc.) do not appear to share this interaction with clopidogrel | |
Rivaroxaban | Antiplatelet agents such as clopidogrel may enhance the anticoagulant effect of rivaroxaban. Avoid concurrent use of clopidogrel with rivaroxaban unless the anticipated benefits outweigh the risks of bleeding. Canadian rivaroxaban labeling recommends avoiding concurrent use with any antiplatelet agent, while the U.S. rivaroxaban labeling recommends caution and increased monitoring if used with any other antiplatelet agent. Any combined use should only be undertaken with increased monitoring for evidence of bleeding | |
Treprostinil | The prostacyclin analogue, Treprostinil, increases the risk of bleeding when combined with the antiplatelet agent, clopidogrel. Monitor for increased bleeding during concomitant thearpy | |
Warfarin | Increased bleed risk may occur as a result of additive anticoagulant effects. Increase monitoring for signs and symptoms of bleeding during concomitant therapy. | |
Food interactions | Take without regard to meals | |
Class effects | Thienopyridines may be slightly more effective than ASA for preventing serious vascular events in high-risk patients (Level 2 [mid-level] evidence) [35] | |
Clopidogrel plus ASA | Related posts: Neuroscience Critical Care: Two Experts’ Point of View
Neurocritical Care of Patients With Arteriovenous Malformations Neuroscience Critical Care: Two Experts’ Point of View
Neurocritical Care of Patients With Arteriovenous Malformations
 Pathophysiology of Intracranial Hypertension
Concepts and Management of Brain Death and Management of Potential Organ Donation Pathophysiology of Intracranial Hypertension
Concepts and Management of Brain Death and Management of Potential Organ Donation
 Extracranial Atherosclerotic Carotid Artery Disease Extracranial Atherosclerotic Carotid Artery Disease
 Acute Paraplegias and Quadriplegias of Non-traumatic Cause Acute Paraplegias and Quadriplegias of Non-traumatic Cause
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |