Applications of Cellular Therapy in Pediatric Neurosurgery
The development of cellular therapy, particularly stem cell (SC) therapy, for the treatment of central nervous system (CNS) disease has progressed rapidly in recent years. Scientists and translational clinicians have targeted SC therapy to broad categories of illness, from inherited metabolic diseases, such as neuronal ceroid lipofuscinosis (NCL), to neoplasms, such as brainstem gliomas. SCs have the potential to revolutionize therapy for previously untreatable CNS diseases. SC therapy, however, is challenged by scientific hurdles as well as by ethical controversy.
SCs have been defined as “…a population of cells capable of indefinite self-renewal that give rise to ‘daughter’ cells committed to specific differentiation lineages through asymmetrical cell division.”1 The term stem cell may refer to tissue derived from embryonic or adult sources, to cells with limited or unlimited potential to divide and replicate, and to cells that are already committed to neuronal, astrocytic, or oligodendrocytic lineages. Asymmetric cell division refers to the ability of a cell to produce a copy of itself plus a more differentiated cell, whereas symmetric division is the ability of a cell to produce two copies of itself.2 Based on the local environment and the presence of various differentiation factors, SCs have the ability to differentiate into any cell type from any of the three germ cell layers. During differentiation, SCs pass through specific, stereotyped stages depending on lineage. For example, during differentiation into a neuron, an embryonic stem cell (ESC) must pass through the following stages: ESC, neuroepithelial cell, neural stem cell/progenitor, and neuron.3,4 Generally, as SC differentiates, its ability to self-renew and multiply diminishes.
The use of SCs in clinical translation is in its infancy. Very few human clinical trials are complete, with even fewer of these studying pediatric patients and diseases. This chapter first reviews the various types of SCs and their potential sources of origin for human therapy. We then contrast cellular therapy with strategies using other biological agents. We briefly outline various methods of delivering cellular therapy to the CNS and then review preclinical and, where available, clinical trials of cellular therapy for brain tumors, epilepsy, stroke, lysosomal storage disorders (LSDs), and spinal cord injury. Finally, we address limitations to the use of cellular therapy in children, including ethical considerations.
7.1 Sources of Stem Cells
7.1.1 Embryonic Stem Cells
ESCs are isolated from the inner cell layer of blastocysts that develop approximately 5 to 6 days following fertilization.1,5 ESCs are derived from the pre- or peri-implantation embryo, have the ability to undergo undifferentiated proliferation, and are capable of pluripotent differentiation into all three cell layers: ectoderm, mesoderm, and endoderm.5 Various ectodermal derivatives of ESCs include dopaminergic neurons6–8 and neural precursors,9 whereas mesodermal derivates include blood vessels.10
Human ESCs may be retained in vitro for years and cultured with a high yield.11,12 They also have the potential for controlled differentiation and subsequent transplantation as neural stem cells (NSCs), astrocytes, or neurons. During normal development, ESCs within the neural plate differentiate based on their reciprocal interactions with surrounding cells and with gradients of morphogenic proteins, including Wnt, Shh, bone morphogenic protein (BMP), and fibroblast growth factors (FGFs).13 These biological variables can be used to direct the differentiation of ESC cultures in vitro for therapeutic purposes.
The use of ESCs for human therapy has been restricted because of their neoplastic potential. Because ESCs have the ability to robustly differentiate into all germ cell layers, teratoma formation has been reported in several animal models.14–16 In another animal model, undifferentiated neuroepithelial cell mitotic activity was found after ESC transplantation.17
At the time this chapter was written, only 152 ESC lines were eligible under U.S. federal regulations for use in National Institutes of Health (NIH)–funded research.18 For regulatory, practical, and ethical reasons, the supply of human ESCs is quite limited. Efforts to develop tools for cellular therapy with biological properties similar to those of ESCs have therefore garnered significant scientific attention in recent years.
Somatic Cell Nuclear Transfer
Somatic cell nuclear transfer (SCNT) involves replacement of the nuclear contents of an oocyte, or egg cell, with those of a somatic donor cell.5 The engineered cell is then artificially activated in a process analogous to fertilization by sperm and undergoes development. Using this technique, Rideout and colleagues replaced the Rag2 gene in transplanted SCNT ESCs, producing allogeneic mature B and T cells in rodents with Rag2 gene deletion, thus restoring their cellular and humoral immunity.19 In other studies, SCNT ESCs increased hemoglobin A protein levels and decreased polychromasia and anisocytosis in a mouse model of sickle cell anemia.20 This approach may be used in the future to treat patients who have known gene deletions or mutations with SCNT ESCs containing normal copies of the specific missing or mutated gene. At the time of this writing, however, SCNT ESCs have not yet been established using a human ovum.
Transcription-Induced Pluripotency
Expression of genes normally active in ESCs can induce adult fibroblasts to enter a developmental state similar to that of ESCs.21 The four genes necessary to create these “induced pluripotent stem cells” (IPSCs) include Oct3/4, Sox2, Klf4, and c-Myc.21 The creation of IPSCs with fibroblasts from nonhuman primates,22 and soon thereafter from humans,23 ignited intense public interest in these cells as an ethically benign alternative to ESCs for human clinical therapy.24
Like ESCs, IPSCs demonstrate germline transmission of their genetic material. Unfortunately, as with other ESCs, a substantial number of the offspring of transplanted animals develop teratomas, possibly because of the activation of c-myc.23 This finding may limit the use of IPSCs in human therapy.
7.1.2 Adult Stem Cells
Adult stem cells (ASCs, sometimes also referred to as somatic stem cells) are multipotent progenitor cells, in distinction to pluripotent ESCs.5 ASCs are isolated from the brain, bone marrow, or other organ systems of humans or animals and can be cultured and expanded in vitro.25,26 Although they are referred to as “adult” SCs, the source of these cells in many cases is organ-specific fetal tissue. ASCs differentiate into cells of other tissue origin only via the process of “transdifferentiation.”27,28 ASCs grown in the presence of Lin28, Nanog, Sox2, and Oct4 exhibit many characteristics of ESCs, including pluripotency.29 Such modified ASCs could be extracted from, modified, and reimplanted into the same patient, potentially avoiding immunologic complications and the ethical concerns arising from the use of ESCs in human therapy.
7.1.3 Primary Neural Cells
Primary neural cells (PNCs) are obtained from fetal cerebral tissue. PNCs cultured from human fetal mesencephalon were used in two clinical trials for the treatment of Parkinson disease (PD).30,31 Advantages of PNCs include a low risk for tumor formation and predictable phenotype.13 However, because they are already differentiated, PNCs largely lack the ability to expand in culture, significantly limiting their human therapeutic potential.
7.1.4 Neural Stem Cells
NSCs may be of embryonic or somatic origin.32 NSCs proliferate within the subventricular zone (SVZ) of mammals, follow a rostral migratory stream, and form new neurons within the olfactory bulb.33–35 NSCs are also responsible for the generation of new dentate gyrus granule cells in the hippocampus.36,37 Frequently, the terms neural stem cell (NSC) and neural progenitor cell (NPC) are used interchangeably. Unlike NSCs, however, NPCs are limited in their ability to self-replicate and typically do not form neurospheres.38
Essentially, all NSCs derive from cells that produce glial fibrillary acidic protein (GFAP)39 and give rise to neurons, glia, and oligodendrocytes.40 A variety of trophic factors direct NSC differentiation into these specific cell lineages. Platelet-derived growth factor (PDGF),41 transforming growth factor-β1 (TGF-β1),42 neurogenin, mammalian AS-C homologue (MASH), and helix loop helix factor (HLH-f)43 facilitate differentiation into neurons. BMP42 and leukemia inhibitory factor facilitate, while Noggin (a BMP inhibitor) interrupts, differentiation into astrocytes.43,44 Finally, insulin-like growth factor-1 (IGF-1) promotes oligodendrocytic differentiation.42 The expression of various transcription factors can similarly influence NSC differentiation.2 Environmental factors also influence differentiation into CNS cell subtypes.45,46 One example is the differentiation of SCs into dopamine-producing neurons necessary for the treatment of PD.26,47
7.1.5 Bone Marrow–Derived and Umbilical Cord Blood Stem Cells
Bone marrow–derived mesenchymal stem cells (BMSCs) are multipotent cells capable of re-creating lineages independent of the hematopoietic system. The key limitation to using BMSCs for treating CNS diseases is technical difficulty in inducing their transdifferentiation. However, BMSCs have the capability to differentiate into all three germ cell layers48 and have been transdifferentated into NSCs.49,50 BMSCs have been shown to cross the blood–brain barrier (BBB), thereby offering the potential for CNS vascular repair following stroke51 and remyelination following white matter injury.52 BMSCs may also have a neuroprotective effect, enhancing host neuronal survival after injury by releasing neurotrophic growth factors.53
Umbilical cord blood (UCB) is also rich in mesenchymal SCs.54 Engraftment rates may be higher for UCB than for BMSCs in treating Hurler syndrome,55 although such differences are not consistent.56 Advantages of UCB include low incidence of graft-versus-host disease, low risk for transmitting disease, rapid availability, and possibly a better graft rate than other hematopoietic SC sources.57
Common sources of SCs are reviewed in ▶ Table 7.1.
| Cell type | Donor source | Advantages | Disadvantages |
| Embryonic stem cells (ESCs) | Embryo | Undifferentiated proliferation Capable of differentiation into all three germ cell layers Long-term in vitro storage | Neoplastic potential Difficulty directing differentiation in vitro |
| Somatic cell nuclear transfer embryonic stem cells (SCNT ESCs) | Oocytes and somatic donor cells | Differentiation potential similar to that of ESCs | Efficacy not yet shown in humans |
| Induced pluripotent stem cells (IPSCs) | Adult fibroblasts plus specific genes | Avoids ethical issues of ESCs Differentiation potential similar to that of ESCs | Neoplastic potential |
| Adult stem cells (ASCs) | Developed organs (e.g., brain, bone marrow) | No immunocompatibility or ethical issues for autograft Expandable in vitro Safe | Restricted neural differentiation potential Questionable functional differentiation |
| Primary neural cells (PNCs) | Fetal cerebral tissue (mesencephalon) | Low neoplastic potential Predictable phenotype | Unable to expand in vitro Require stage-specific embryonic source Limited differentiation potential |
| Neural stem cells (NSCs)/ Neural progenitor cells (NPCs) | Embryo Subventricular zone of adults | Able to differentiate into neurons, glia, and oligodendrocytes Long-term vitro storage and expansion | Limited sources |
| Bone marrow–derived stem cells (BMSCs) | Bone marrow | Capable of differentiation into all three germ cell layers Crosses the blood–brain barrier Neuroprotective effects | Difficulty with transdifferentiation into NSCs |
| Source: Adapted from Guillaume DJ, Zhang SC. Neuronal replacement by transplantation. In: Bottenstein J, ed. Neural Stem Cells: Development and Transplantation. Norwell, MA: Kluwer Academic Publishers; 2003:309. | |||
7.2 Contrast to other Biological Agent Strategies
Cellular therapy follows a long line of rationally designed and targeted therapies for incurable and previously untreatable neurodegenerative diseases. This era of designed therapies began with the development of L-3,4-dihydroxyphenylalanine (L-DOPA) for the treatment of PD.58 Although L-DOPA relieved some of the symptoms resulting from the degeneration of nigral dopaminergic neurons in patients with PD, it did not interfere with the inexorable underlying pathologic process. Subsequent developments in neurodegenerative disease therapy reflect both an attempt to more directly interrupt disease pathophysiology and growth in basic biological expertise in protein chemistry, genetics, and ultimately cellular therapy. Development of novel therapies for the incurable, inherited LSDs provides an example of this progress. The LSDs are characterized by the deficiency of an enzyme or enzymes that results in the accumulation of pathologic material in lysosomes, ultimately causing neuronal death and CNS dysfunction.
7.2.1 Enzyme Replacement Therapy
Intravenous (IV) enzyme replacement therapy (ERT) is the principal approach for treating many inherited enzyme deficiency diseases, including the LSDs59 mucopolysaccharidosis (MPS) VI (Maroteaux-Lamy syndrome),60 glycogen storage disease type 2 (Pompe disease),61 MPS I (Hurler syndrome),62 α-galactosidase A deficiency (Fabry disease), and glucocerebrosidase deficiency (Gaucher disease).63
Hunter syndrome is caused by a deficiency of the lysosomal enzyme iduronate-2-sulfatase. IV infusion of idursulfase for Hunter syndrome has resulted in multiple somatic improvements, such as increased mobility, decreased hepatomegaly, and decreased respiratory infections.59 IV delivery of idursulfase has not, however, resulted in similar improvements in CNS function because of the relative failure of IV therapy to deliver replacement enzyme to the CNS.64 In many disease states, ERT is completely ineffective because of BBB obstruction of CNS penetration.63
7.2.2 Viral Vector Gene Therapy
The NCLs are examples of diseases untreatable with peripheral ERT. Late infantile NCL, an autosomal-recessive lysosomal storage disorder caused by mutation of the CLN2 gene, results in profound deficiency of the lysosomal storage enzyme tripeptidyl peptidase1 (TPP1), neuronal loss, progressive neurologic decline, and death.65 Adeno-associated viruses (AAVs) can transfer human CLN2 cDNA to genetically deficient cells in vitro and in rodent and primate animal models.66 Gene transfer therapy increased levels of the deficient enzyme, TPP1, in the striatum, substantia nigra, frontal cerebral cortex, and thalamus of the injected hemisphere, and in the contralateral frontal cerebral cortex. Another group found similar results in an animal model of a related disorder, infantile NCL.67
In a study of 10 children with late infantile NCL, AAV vector gene therapy may have slowed disease-related brain atrophy and neurologic deterioration compared with control subjects.65 Given the low numbers of patients in this study, no difference in outcomes was statistically significant. Several adverse events were noted, including the death of one patient secondary to postoperative status epilepticus, as well as development of a humoral immunoresponse to the vector in four patients.
Gene replacement therapy is also being tested in animal models of other LSDs, including MPS VII (Sly syndrome) and MPS IIIB (Sanfilippo syndrome). In rodent models of MPS VII, direct intracerebral injection of the AAV2 vector, transplantation of retrovirus-transduced fibroblasts, and transplantation of adenovirus-transfected amniotic epithelial cells all reduced pathologic lysosomal storage.68–70 A similar viral gene therapy approach has been effective in a rodent model of MPS IIIB.71 This viral therapy strategy may be improved by using organ-specific NPCs.72,73
Viral gene therapy for neurodegenerative disease faces a number of ongoing challenges. Individual viral vectors and genetic payloads must be designed for every genetic disease and disease variant. Each of these vectors must undergo extensive safety testing and regulatory approval.74 The efficiency of transfection may be limited in many cases, and more efficient viruses may also pose a higher risk for associated pathologic inflammatory responses.75 Finally, although they may spread by transfection, viruses have no inherent migratory potential. Thus, the delivery of the therapeutic agent to the entire expanse of the human nervous system in LSD is a daunting challenge and would likely require vascular delivery across the BBB.
7.3 Clinical Applications of Stem Cell Therapy
The inherent properties of SCs may prove advantageous in overcoming the daunting challenges of treating CNS neurodegenerative disease. For example, implanted lineage-specific, or adult, SCs may survive indefinitely in the host environment, self-replicate or expand, and migrate significant distances within the CNS.76,77
For LSDs and other enzyme deficiency diseases related to an isolated genetic defect, SCs may be used in a cross-correction strategy. In this circumstance, transplanted, genetically normal SCs produce and secrete the missing protein, which diffuses locally and supplies the genetically deficient host cells (▶ Fig. 7.1).13
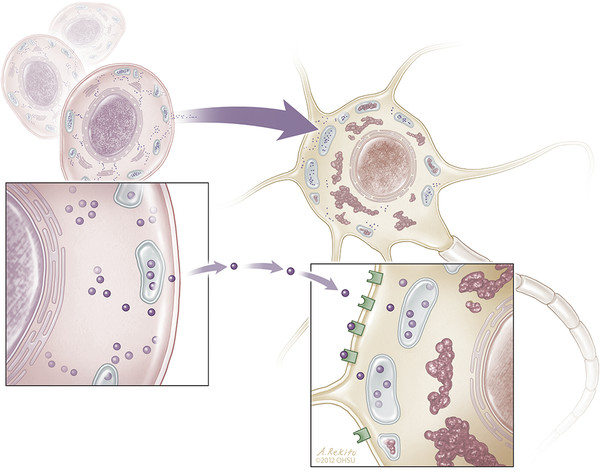
Fig. 7.1 Intercellular transfer of deficient enzymes from an intracereberally transplanted neural stem cell to a neuron in a process known as metabolic cross-correction. Stem cells secrete active enzymes, which are then taken up by receptor-mediated endocytosis into enzyme-deficient neurons and transported to lysosomes, reducing precursor accumulation, normalizing lysosomal function, and protecting neurons from degeneration.13
NSCs are capable of differentiating in situ into the three major cell lineages of the mature CNS: neurons, astrocytes, and oligodendroglia78 (▶ Fig. 7.2). The most sophisticated and technically challenging use of SC therapy for CNS diseases will be replacement of damaged neuronal circuits and glial architecture based on these regenerative and pluripotent properties. Cross-correction models, however, do not require lineage-specific differentiation or circuitry repair.
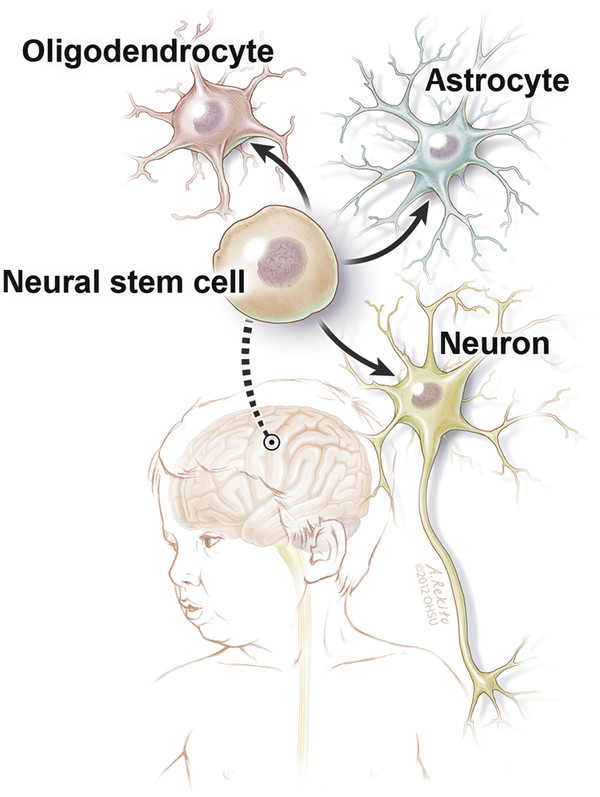
Fig. 7.2 Schematic diagram of an intracerebrally transplanted neural stem cell’s capability to differentiate into an oligodendrocyte, astrocyte, or neuron.
7.3.1 Cellular Delivery
Various methods for delivery of SCs to their therapeutic target have been employed, including IV, intra-arterial (IA), and direct surgical implantation. IV administration of SCs is less invasive than either IA or direct surgical implantation, but delivery of cells to the CNS is significantly hindered by a pulmonary first-pass effect,79 with only approximately 4% of cells entering the brain.80 Up to 21% of cells injected into the internal carotid artery enter the brain,81 but this technique may be complicated by the occurrence of microemboli.82 Direct intracerebral transplantation followed by intraventricular and IV transplantation results in the greatest number of SCs at the site of a CNS lesion.83 However, functional recovery does not necessarily correlate with the absolute number of viable transplanted cells into the targeted area.84
Implanted SCs have the ability to improve neurologic outcomes without crossing the BBB. In several preclinical stroke models, neurologic function improved despite that fact that few or no SCs were found in the brain.80,85 This is likely due to the ability of NSCs to counteract inflammatory cytokines, toxic metabolites, free radicals, and excitotoxins released systemically after an insult.86 NSCs also produce multiple protective factors such as glial cell line–derived neurotrophic factor (GDNF), which may contribute to rescuing dopaminergic neurons in PD1 and improving functional outcomes after stroke,87 as well as glial and neural growth factors, such as neurotrophin-3 (NT-3) and ErbB-2, respectively.88,89 Other neuroprotective factors enhanced by NSCs include fibroblast growth factor (FGF), vascular endothelial growth factor (VEGF), bone-derived neurotrophic factor (BDNF), nerve growth factor, and ciliary neurotrophic factor.87,90
7.3.2 Disease States
Ischemia
Cerebral ischemia from moyamoya disease, genetic disorders, and perinatal thromboembolic disease results in profound morbidity in the pediatric population. Cerebral ischemia triggers the proliferation of NPCs in the SVZ91,92 and dentate gyrus. NPCs migrate to areas of focal ischemia in rodent models.93,94 SVZ cells may have the capacity to regenerate some striatal neurons following ischemia.95 Migration of NSCs to the site of ischemic injury is activated by erythropoietin.93,96,97 Epidermal growth factor receptor (EGFR) levels in the SVZ increase following cerebral ischemia, while infusion of EGF into ischemic brain increases the number of EGFR-positive neuroblast precursors,95 which migrate into the striatum and cerebral cortex following ischemic insults to those regions.93,98 Although the therapeutic implications of these findings remain unclear, it appears that SVZ and other stem and progenitor cells demonstrate tropism toward sites of CNS injury.
For neuronal circuit repair after stroke, implanted SCs must migrate to appropriate targets and also differentiate into appropriate cell types. In a rodent ischemia model, implanted ESC-derived NPCs differentiated into neurons and glia with electrophysiologic function.99 Similarly, transplanted NSCs in a gerbil model of ischemia yielded NeuN-positive neurons and glial fibrillary acidic protein (GFAP)–positive glia, grew synaptic connections between host and graft tissue, and improved neurologic function.100 After intraventricular injection, NSCs in a mouse model of neonatal hypoxic–ischemic encephalopathy migrated to the site of ischemic injury and differentiated into neurons, resulting in improved memory and learning compared to controls 8 months after transplantation.101 Intracerebral transplantation of NSCs in a rodent model of cerebral palsy (CP) yielded a 5% population of NSC-derived neurons in the infarction cavity.102 At the time of this writing, there were no published clinical trials of SC therapy for CP; however, two clinical trials have been completed, and three trials are currently recruiting patients.103 When the NSC neurons were transduced with a retroviral neurotrophin (NT-3) before transplantation, the proportion of NSC-derived neurons in the infarction cavity increased to 20% and in the penumbra to 80%, raising the possibility that these therapies interact synergistically. In other studies, IA104,105 or IV84 SC injection to treat stroke has also improved neurologic recovery.84
Human studies support the potential of SC therapy for the treatment cerebrovascular accidents (CVAs). Human neuronal cells implanted into 14 adult patients with ischemic or hemorrhagic CVA were associated with improved activities of daily living and memory compared to baseline, but not compared to nonimplanted controls.106 No significant change was noted in European Stroke Scale motor score. In one patient, who expired 27 months after transplantation, human neuronal cells were found adjacent to the lacunar infarct.107 Another clinical study of intracranial implantation of fetal porcine cells for stroke therapy was terminated by the U.S. Food and Drug Administration (FDA) after enrolling only five patients because of the occurrence of adverse events, including worsened motor deficits and seizure onset following transplantation.108
SCs may play different roles in stroke therapy depending on the timing and route of administration. SC administration very early after ischemic stroke may provide neuroprotection of salvageable pneumbra by increasing neuronal proliferation and abating edema,102,109 although the proportion of SCs surviving transplantation is relatively low.76,110 By contrast, SCs administered peripherally 21 days after ischemic injury survive in greater numbers and differentiate via neuronal and glial lineages within injured brain.111
Tumors
Currently, the most common application of SC therapy in children is high-dose chemotherapy (bone marrow–ablative) with autologous hematopoietic SC transplant rescue as a salvage therapy for recurrent or high-risk brain tumors. The most common source of SCs is peripheral blood, followed by bone marrow. This treatment has been used in a variety of neoplasms, including medulloblastoma, malignant glioma,112 ependymoma, germ cell tumors, and atypical rhabdoid teratoid tumors.113 Responses are variable (0 to 75% long-term survival) and often at the price of substantial toxicity, including death in up to 33%, mucositis, neutropenia, and renal and hepatic failure.113 There are currently approximately 40 registered SC-based clinical trials worldwide for pediatric brain tumors, which will continue to define the safety and efficacy of this treatment strategy over time.114
Brainstem gliomas and supratentorial malignant gliomas constitute approximately 15%115 and up to 10%,116 respectively, of pediatric brain tumors. Neural SCs appear to have a high affinity, or tropism, for brain tumor tissue, making them ideal vehicles for the introduction of therapeutic enzymes and chemotherapy.113,114 Bone marrow–, adipose tissue–, and umbilical cord–derived SCs all show tropism for brain tumor tissue, with comparable rates of SC survival and percentage of migratory cells.115 Tumors attract SCs via the secretion of numerous chemoattractant factors, including cytokines, VEGF, stromal cell–derived factor-1 (SDF-1), NT-3, TGF-β1, and interleukin-8 (IL-8).114,117
Interleukin-4 (IL-4) cDNA delivered via SCs in a rodent model of malignant glioma resulted in tumor regression, as determined by magnetic resonance (MR) imaging, and significantly improved survival.118 In this study, treatment with unaltered NSCs alone also provided a degree of intrinsic antitumor activity. In another study, BMSCs expressing interferon-γ (IFN-γ) increased survival in a model of supratentorial malignant glioma.77
Mesenchymal SCs (MSCs) have also successfully delivered prodrug converting enzymes in animal models of brainstem glioma. As a proof of concept, human NSCs engineered to express cytosine deaminase, a prodrug enzyme, and IFN-γ, an antiangiogenic and immune response enhancer, migrated, increased tumor cell apoptosis, and substantially reduced tumor volume in a model of brainstem glioma.115 Human adipose tissue–derived MSCs genetically altered to convert the prodrug of irinotecan, a topoisomerase-1 inhibitor, into the active form significantly extended the survival of rats treated with both the engineered MSCs and irinotecan.119 This strategy is also being tested in human clinical trials. Genetically modified NSCs expressing an enzyme for the conversion of 5-fluorocytosine (5-FC) to 5-fluorouracil (5-FU) are being evaluated for the treatment of recurrent high-grade gliomas.120
NSCs also show substantial tropism toward medulloblastoma. Even when injected in the contralateral hemisphere in an animal model, human NSCs engineered to convert systemic 5-FC to 5-FU reduced the size of medulloblastoma tumors by 76%.121 In another model, human NSCs engineered to convert irinotecan to its active form reduced the growth rate of cerebellar tumors.122
Lysosomal Storage Disorders
The LSDs are a class of over 40 disorders,123 each defined by “…a specific inherited enzyme deficiency that leads to accumulation of complex macromolecules, such as sphingolipids, glycogen, mucopolysaccharides, and glycoproteins in lysosome[s],” ultimately resulting in neuronal death.64 Many of the LSDs have their most devastating effects on the CNS. Because of this, the transmission of peripherally delivered therapies, such as enzyme replacement, across the BBB has posed a significant impediment to therapy. For many LSDs, which are often severely morbid or fatal, there is no effective treatment. SC therapy for LSD offers the potential to deliver replacement enzymes in a sustainable fashion within the CNS by using a cross-correction model.
Intraventricular delivery of enzyme, although technically challenging, can positively impact some LSDs. In rodent models of the GM2 gangliosidoses (Tay-Sachs disease and Sandhoff disease), intraventricular injection of recombinant human HexA (Om4HexA) with a high mannose-6-phosphate (M6P)-type-N-glycan content reduced levels of sphingolipids, GM2, and asialo-GM2 (GA2) in brain parenchyma and improved both motor function and survival.124
SCs may provide continuous and potentially durable enzyme replacement for LSDs. To date, however, the clinical benefits of SC therapy for LSD have been mixed, and success in preclinical models has not necessarily translated to patients. In a rodent model of Niemann-Pick disease, NSCs implanted into the cerebellum increased survival but did not affect weight loss or neurologic function.125 A number of animal studies in which multiple treatment methods were used in a single animal, such as BMSC transplantation and ERT, have shown outcomes superior to those obtained when either therapy was used individually.126 Multimodality therapy may also prove useful in human patients.
MSCs delivered to 12 patients with metachromatic leukodystrophy or Hurler syndrome did not improve cognitive or physical development.127 UCB transplantation in 25 infants with Krabbe disease improved neurocognitive function and development in those patients who were asymptomatic at the time of transplant.128 Similar results were found after UCB for X-linked adrenoleukodystrophy (X-ALD).129 Hematopoietic SC transplantation is therefore currently advised for use only in asymptomatic patients, creating a premium for the early perinatal diagnosis of inborn errors of metabolism.57
Intracerebral transplantation of human NSCs in a rodent knockout model of infantile NCL resulted in SC engraftment, migration, production of the missing palmitoyl–protein thioesterase 1 (PPT1) enzyme, reduced burden of autofluorescent material, and neuronal preservation in cortex and hippocampus.78 Based on these results, a Phase I trial of intracerebral transplantation of human NSCs for infantile NCL and late infantile NCL was recently completed. This study utilized multiple intracerebral and bilateral intraventricular surgical implantations (▶ Fig. 7.3) of very high volumes of SC suspension, with a total dose of SC delivery unprecedented in previous human therapy (up to 1 billion cells per patient). The trial also represented the first human treatment with a purified and characterized NSC, the first human CNS implantation of a purified SC population, and the first surgical CNS transplantation carried out in children.130,131 Six children underwent transplantation and were treated with 1 year of immunosuppression. No surgical, SC-related, or immunosuppressive toxicity was observed, and human leukocyte antigen (HLA) antigenic evidence suggested successful engraftment and migration more than 2 years after transplantation and well after the cessation of immunosuppression. The study was not powered to assess efficacy.130
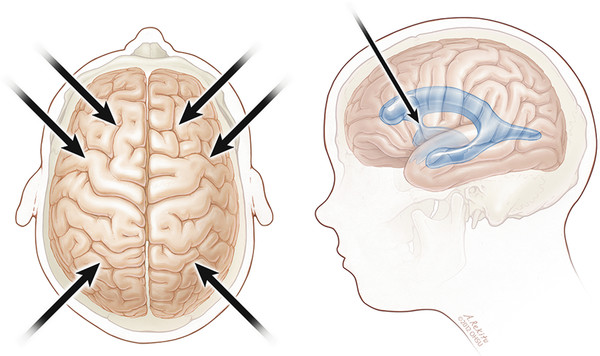
Fig. 7.3 Transplantation of neural stem cells into six intraparenchymal supratentorial sites and bilateral frontal intraventricular sites.
Traumatic Brain Injury
Traumatic brain injury is responsible for nearly 500,000 emergency department visits each year by children ages 0 to 14 years.132 Mortality and morbidity from severe traumatic brain injury have not improved dramatically over the past 30 years, suggesting the need for novel treatment strategies for neuroprotection after primary brain injury and for neuro-restoration.133,134 SC therapy has the potential to deliver these benefits to patients with traumatic brain injury.
Injured brain parenchyma expresses an abundance of inflammatory mediators, creating a hostile host environment for transplanted SCs. Implanted SC viability correlates with the degree of injury.135 As in stroke, SC administration is most likely optimal immediately following the injury, before the peak inflammatory response, or after the acute inflammatory process has subsided.82
Bone marrow–derived SCs administered intracerebrally,136 IV,50,137 or IA138 after traumatic brain injury all survived for up to 3 months and migrated to the injury site. NSCs have also been found to migrate to injury sites and are associated with improved neurologic function, surviving up to 14 months.82,139 UCB administered after traumatic brain injury contains SCs that differentiate into neurons and astrocytes and result in improved function.140
In a preliminary study, 10 children with severe traumatic brain injury received IV autologous bone marrow–derived mononuclear cells within 48 hours of injury.141 This feasibility study found no evidence of treatment-related CNS or systemic toxicity but was not designed to assess neurologic or cognitive functioning.
Epilepsy
An increasing amount of preclinical research has focused on using NSCs to treat mesial temporal lobe epilepsy and, to a lesser degree, epilepsy related to cortical dysplasia. The microenvironment of the epileptogenic hippocampus is favorable to the implantation and integration of SCs.142 Lack of inhibition at the dentate hilus allows the propagation of ictal discharges from the entorhinal cortex to the hippocampus and subsequently the rest of the brain.143 One strategy used for the treatment of refractory epilepsy is therefore transplantation of inhibitory NSCs.144 The dentate gyrus may be the most appropriate target for inhibitory precursor transplantation. Other treatment strategies using SCs for epilepsy include promotion of surround inhibition and neuronal replacement.145
Transplanted SCs require differentiation of NSCs into inhibitory neurons—for example, those utilizing γ-amino butyric acid (GABA) neurotransmission—to cause mesial temporal inhibition. NSCs implanted into the dentate gyri of rats produce GABA-ergic cells, raise seizure thresholds, and shorten seizure duration.146 Transplanted embryonic medial ganglionic eminence cells engraft and migrate in large proportions to the hippocampus, striatum, and neocortex in animal models of partial and generalized epilepsy, with prolonged survival.147–150 Transplanted cells also influence host neuronal circuitry by increasing levels of synaptic inhibition in neocortical pyramidal neurons.147 Loss of inhibitory interneurons likely also plays an important role in the pathogenesis of cortical dysplasia, which may represent a potential target for NSC-derived inhibitory neuron transplantation.151,152
Cellular therapy could also reduce seizures in mesial temporal lobe epilepsy by replacing dysfunctional or damaged neurons. NPCs implanted into the cerebral cortex and hippocampus of rats develop projections to the host thalamus and contralateral hippocampus, generate action potentials, and harbor N-methyl-D-aspartate (NMDA)–mediated excitatory and GABA-mediated inhibitory postsynaptic currents.153,154
Spinal Cord Injury
Like the brain, the zone adjacent to the spinal cord ependymal lining is a key site in for NSC formation and proliferation after injury.155,156 The host environment at the site of spinal cord injury is important to potentiate SC-induced recovery. Following the clearance of necrotic debris, gliotic scarring inhibits the regeneration of severed axons.157 Implantation of selected macrophages may play a role in preventing a glial scar, clearing cellular debris, and promoting the migration of endogenous SCs to the site of injury.158 Recovery of motor function in an animal model is significantly increased when SC transplants and neurotrophins are administered 2 to 4 weeks following spinal cord transection compared to those administered more acutely, in the presence of deleterious, acute inflammatory responses to injury,159 including such mediators as tumor necrosis factor-α (TNF-α), IFN-γ, and IL-β1.160
In animal models, direct implantation of SC into the injured spinal cord dramatically improves function.161,162 NSCs engrafted into the injured cord tend to stay confined to the white matter and differentiate into glial cells,163 particularly oligodendrocytes that form myelin.164 Nevertheless, those NSCs that differentiate into astrocytes165 and neurons166 also have the potential to improve neurological function. Implantation of UCB into the injured spinal cord has also produced myelin167 and improved neurological function.168
In a Phase I clinical trial,169 the injection of autologous macrophages into the spinal cords of eight patients with complete spinal cord injury was associated with recovery in three patients from American Spinal Injury Association Impairment Scale grade A to grade C. Phase II studies are currently under way utilizing this technique.170 Patients with both subacute and chronic spinal cord injury treated with BMSCs showed modest functional improvements, particularly when cells were injected into the vertebral artery.171 PNCs of fetal origin were directly injected at the site of injury in patients with postinjury syringomyelia without adverse events and with MR imaging correlates of successful engraftment.172
7.4 Challenges to Cellular Therapy
7.4.1 Biological Challenges
Immunocompatability
ESCs differ genetically from recipients but express little HLA class I and no class II.173 As ESCs differentiate, class I major histocompatibility complex is upregulated, so that immunosuppressant therapy is required after transplantation.174 Alternatively, it may be possible by using nuclear transfer or other techniques to create donor ESCs genetically identical to the recipient.175,176 The use of immune-inert, semipermeable capsules to house SCs may also promote immunocompatibility and protect SCs from attack by inflammatory modulators resulting from direct cerebral implantation.131
The proper dose, timing, and duration of immunosuppression after CNS SC transplantation are controversial, especially given the medical fragility of the population of patients for whom SC therapy is currently being explored. In two studies of transplantation of human fetal mesencephalic tissue for PD, one study used immunosuppression and the other did not.30,31 Grafted cells were present on postmortem histology from patients in both studies, although an activated macrophage marker (CD45) was increased around SC graft injection sites in nonimmunosuppressed patients.30 In a Phase I trial of human NSC transplantation for NCL, patients were treated with immunosuppression just before and for 1 year after implantation. Frequent medication adjustments were required because of the interaction of anticonvulsant and immunosuppressant drugs, but there were no obvious additional adverse effects. Evidence for persistent engraftment was present in two of three postmortem examinations, in one case more than 2 years after transplantation.130 In a single patient with stroke in whom human neuronal cells derived from a teratocarcinoma line were implanted and who received peritransplantation immunosuppression for 9 weeks, the implanted neurons survived for more than 2 years.107
Neoplastic Potential
ESCs have many similarities to neoplastic cells, including the ability to undergo unlimited, undifferentiated proliferation and to form tumors in animal models of PD.15,16 ESCs readily differentiate into all three germ layers and have a propensity to form teratomas. Cell overgrowth14,17 and metastatic retinoblastoma formation have also been observed in animal models.177 To reduce their oncologic potential, ESCs must be used in lineage-specific, differentiated form, such as NSCs. However, brain tumor formation was reported in a human patient 4 years following implantation of fetal NSCs.178 Pluripotent SCs may be sorted according to their identifying cell surface proteins13 or by leveraging cell type–specific transcription factors.179
Other Biological Obstacles
Scientists and clinicians must overcome several additional obstacles before SC therapy can be widely used in the treatment of pediatric neurologic disorders. First, there is currently a lack of clinical markers that specifically identify transplanted NSCs in order to distinguish them from other progenitor or differentiated host cells.1,2 Second, therapeutic strategies must mitigate host factors. For example, direct intracerebral implantation may affect the viability and functionality of SCs because of the creation of a hostile inflammatory microenvironment.30,31,106,108,130 The underlying disease state may additionally result in gliosis or vacuolization that can adversely affect engraftment, migration, or the function of transplanted SCs. Third, in many disorders, particularly developmentally relevant disorders in childhood, outcomes assessment may be confounded by a lack of appropriate and validated instruments. For example, most validated measures of neurocognition require intact visual and motor functional skills, which are often damaged early (e.g., in LSDs).131 Fourth, it is difficult to track the location and assess the viability of implanted SCs in human patients. Guzman and colleagues used iron nanoparticles to successfully trace migration patterns as assessed by MR imaging and histology.180 Notably, these iron nanoparticles did not affect cellular migration, survival, differentiation, or electrophysiologic characteristics. This technology has the potential to identify single cells.181 Others have successfully employed MR spectroscopy182 and positron emission tomography (PET)183,184 to identify NPCs in humans. Engineering of SCs to facilitate in vivo imaging, however, creates additional regulatory hurdles for their use in human trials.
7.4.2 Regulatory and Ethical Challenges
The regulatory issues surrounding SC transplantation are complex and continue to evolve. In the United States, the FDA oversees the use of SCs and other biological therapeutics in human patients. Because they are human cells that are highly processed and “utilized for other than their normal function,”185,186 SCs must conform to both Section 351 regarding biological products187 and Section 361 regarding human cells and associated products188 of the Public Health Service Act.185 The U.S. Code of Federal Regulations (CFR) Part 1271 details the regulation of “human cells, tissues, and cellular and tissue-based products.”186 Generally, this code was designed to “create a unified registration and listing system for establishments that manufacture human cells, tissues, and cellular and tissue-based products (HCT/Ps) and to establish donor eligibility, current good tissue practice, and other procedures to prevent the introduction, transmission, and spread of communicable diseases by HCT/Ps.”186 Thus, the regulatory standards and checks for the manufacturing of biological material for human use are stringent and expensive.185
SC research faces additional serious challenges related to ethical concerns about the creation and use of human embryonic and other cellular tissue. Both sides in this debate cite concern regarding the value of human life as a motivating factor. Proponents of SC therapy identify concern for patients with severe or fatal, untreatable nervous system diseases as a factor justifying this work. Opponents cite concern that embryonic tissue, representing human life, will be arbitrarily created or destroyed for the purposes of SC therapy. In both cases, sincerely held views deserve the attention and respect of the medical and scientific communities.189
The ethical attention of the public and press has focused to a large degree on ESC transplantation.190,191 In many cases, however, lineage-specific, or “adult,” SCs are actually sourced from fetal tissue. As discussed previously, SCNT and IPSC technology may in future provide a relatively ethically neutral source of human SCs for therapeutic purposes.
The public focus of attention on the ethics of ESC transplantation has diverted necessary attention from a number of other important considerations: Who should own the intellectual property and biological material rights for cells necessary for human therapy? Is the use of “sham” surgery in human clinical trials of cellular transplantation ethical?31 What are the boundaries for the appropriate use of complex and expensive transplantation therapy (particularly in cases in which such therapy may be only palliative)? How can we guard the interests of children and neurocognitively disabled patients enrolled in trials of SC therapy, without entirely denying their access to such therapies and preventing progress in the treatment of their diseases? Because of these and other questions, progress in human SC therapy must occur within the bounds of careful and public regulatory oversight and ethical guidelines.189
7.5 Summary
Stem cells are inherently capable of self-replication and expansion, engraftment, long-term survival in a host environment, and extensive migration. In various circumstances, they can be used to supply missing proteins, provide neuroprotection and trophic support to host cells, or even reconstruct missing or damaged neural architecture.
Significant challenges to successful human therapy with SCs remain, including overcoming barriers to CNS administration, modulating the host environment and immune response, and accomplishing in vivo the tracking of transplanted SCs. Recent scientific developments hold promise for mitigating ethical concerns about and practical limitations on sources of SCs for human therapy.
Pearls
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


