B. Response to medications. Absence of benefit from adequate dosages of dopaminergic drugs, especially levodopa, casts doubt on the diagnosis of IPD and suggests a diagnosis of secondary causes of parkinsonism or one of the Parkinson-plus syndromes. Equally important is determining whether, early in the illness, these medications produced psychiatric side effects such as hallucinations or autonomic symptoms such as severe orthostatic hypotension. The former suggest the possibility of dementia with Lewy bodies (DLB), and the latter indicate possible multiple-system atrophy (MSA). In IPD, psychiatric and autonomic side effects from dopaminergic drugs are not uncommon but usually appear when the illness is at least moderately advanced.
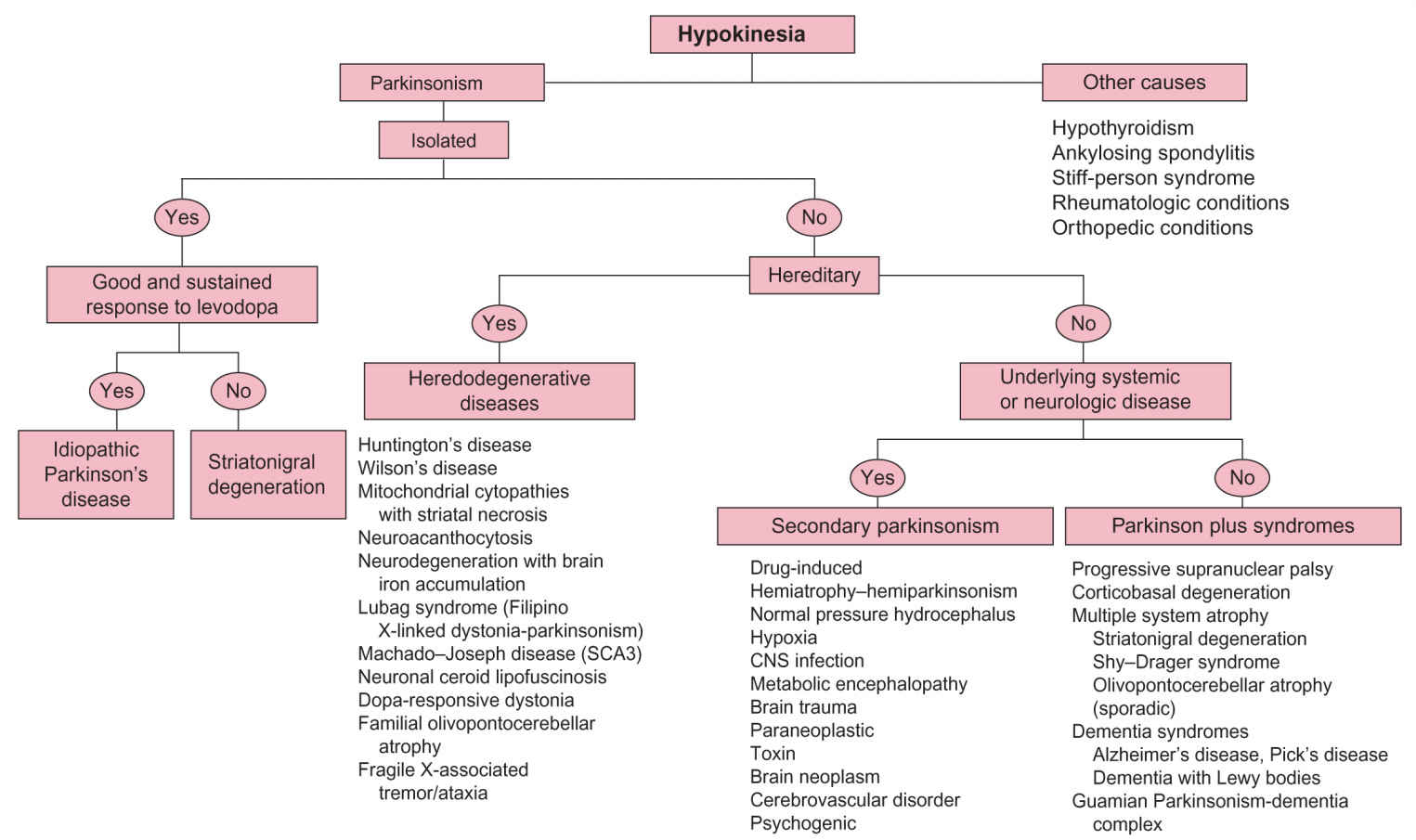
FIGURE 30.1 Algorithm for differential diagnosis of hypokinesia.
C. Cognitive symptoms. Even early in the course of the disease process, patients with IPD may have mild executive and visuospatial dysfunction. Dementia in IPD is largely related to cortical -synuclein deposits in the form of Lewy bodies and Lewy neurites, but cortical amyloid can also contribute to an already impaired patient. Frank dementia is more common among older patients and usually after the illness is moderately advanced. Mild to moderate cognitive symptoms are present in most of the Parkinson-plus syndromes but are seldom the presenting symptom. Severe, early cognitive abnormalities may indicate a primary dementing disorder such as Alzheimer’s disease (AD) or vascular dementia.
D. Psychiatric symptoms. Symptoms suggestive of depression or anxiety may precede the onset of IPD. If hallucinosis (typically visual) is present, determine if it began early or late in the course of the illness and whether it appeared in response to the institution or escalation of an anti-Parkinson drug. Very early appearance of hallucinations in cases of parkinsonism or their presence in an untreated parkinsonian patient raises the probability of DLB.
E. Sleep disorders. The rapid eye movement behavior may precede the onset of parkinsonism by several years in IPD or DLB. Restless legs syndrome and/or periodic limb movements of sleep may be associated with IPD. Discomfort because of rigidity and inability to turn in bed can cause sleep fragmentation (see Chapters 9 and 60).
F. Dysautonomia. Constipation, urinary urgency, impotence, and orthostasis may accompany or even precede IPD. When prominent early, and especially if severe, these symptoms may suggest MSA.
G. Medication usage. Patients must be asked if they are currently taking or have recently received antidopaminergic drugs such as neuroleptics, reserpine, or metoclopramide. In addition, any history of illicit drug use should be ascertained.
H. Family history. IPD has a complex and multifactorial etiology. Patients with Mendelian pattern of inheritance constitute a small minority of the overall Parkinson’s disease (PD) population. Heritable disorders that can mimic PD include Wilson’s disease (autosomal recessive), juvenile Huntington’s disease (HD; autosomal dominant), and essential tremor (ET; autosomal dominant with variable penetrance).
I. Toxic exposure. Exposure to toxins such as manganese or carbon monoxide must be ascertained because both can result in parkinsonism. Less common causes include mercury, carbon disulfide, methanol, and cyanide.
PHYSICAL EXAMINATION
A. The clinical findings of parkinsonism.
1. Hypomimia is characterized by diminished facial expression with infrequent eye blinking. A fixed facial expression, often seen in progressive supranuclear palsy (PSP), consists of an unchanging expression such as surprise in which the forehead may be furrowed, the eyelids retracted, and the nasolabial folds deepened. Myerson’s sign, present in IPD and a variety of other basal ganglia disorders, consists of persistent reflex eyelid blinking to repetitive finger taps applied to the glabella, instead of the normal rapid habituation after the fourth or fifth tap.
2. Hypophonia is characterized by diminished amplitude and inflection of speech. Tachyphemia is an excessively rapid speech pattern, which is a common accompaniment of hypophonia, making such speech even more unintelligible.
3. Rigidity may be predominant in axial muscles (e.g., neck or trunk), in the limbs, or equally severe in both. Increased resistance to passive movement of the involved body part is easily appreciated when rigidity is severe. When subtle, rigidity can be reinforced by asking the patient to alternately open and close the fist of the hand on the side opposite of the arm or leg being tested. The presence of tremor in the same limb demonstrating rigidity gives rise to a rachet-like sensation referred to as cogwheel rigidity.
4. Tremor may appear in one or more forms in patients with parkinsonism.
a. Resting tremor is the hallmark of IPD. Its absence casts some doubt on the diagnosis but certainly does not rule it out. It is also present in some other forms of parkinsonism. The tremor is most commonly seen in the hands and to a slightly lesser extent in the lower extremities and mandible. Rest tremor rarely involves the head and never affects the voice. It appears at a frequency of 4 to 5 Hz and is often at least temporarily extinguished by volitional movement. A subtle tremor can be uncovered by asking the patient to perform difficult mental arithmetic, a mildly stressful task.
b. Action tremor may also be present in IPD as well as in other parkinsonian syndromes, especially those associated with cerebellar dysfunction. It can be present as a postural tremor while the arms are outstretched in front of the patient or as a kinetic tremor while the patient is performing a task such as the finger-to-nose test. Postural tremor alone, in the absence of parkinsonian signs, suggests a diagnosis of ET.
c. Positional tremor. Some tremors are particularly prominent when the involved body part is placed in a specific position. The wing beating tremor of Wilson’s disease is an example of this phenomenon. This tremor is noted when the arms are abducted at the shoulders while flexed at the elbow.
5. Bradykinesia can be documented by simply observing the speed, amplitude, and amount of ordinary movements made by the patient such as gestures or shifting of body position. Repetitive motion tasks such as tapping the index finger against the thumb demonstrate slowness of movement and a progressive loss of amplitude.
6. Impairment of automatic movements is noticeable as a decrease in gesticulation and head movement during conversation, a reduction in the automatic repositioning of limbs while sitting in a chair or reclining in bed, and as a decrease in the amplitude of arm swing while walking. In severe hypokinesia, the affected arm(s) may not swing at all, but rather be held in a semiflexed posture across the trunk.
7. Impairment of repetitive movements such as handwriting or buttoning a shirt is not only performed slowly, but the amplitude of each successive movement typically becomes progressively smaller. This may account for the progressively smaller letters (micrographia) seen when a hypokinetic patient is asked to write a long sentence.
8. Impaired initiation of movement is manifested by difficulty in arising from a chair or hesitancy in taking the first step while attempting to walk. Many patients with IPD have difficulty initiating two motor acts simultaneously such as standing up and shaking hands. Rising from a chair is tested by asking the patient to rise with arms crossed in front of the body to prevent pushing off. The patient may require several attempts to succeed or may be totally unable to arise without using his arms. If the patient is unable to rise without assistance, a judgment must be made as to whether the cause is weakness (which can be tested independently).
9. Gait and posture should be evaluated by having the patient walk a distance of at least 20 feet in an area free from obstacles. Parkinsonian patients often display reduced stride length and arm swing, stooped posture, difficulty in initiating gait, and turns with the body moving as a single unit (en bloc). In more advanced cases, progressively more rapid, small steps as the body leans forward (festination) and “freezing” in midgait may be observed.
10. Freezing is a sudden involuntary cessation of a motoric act, usually walking, while other functions remain intact. This phenomenon is confined to basal ganglia disorders. It may occur spontaneously or may be provoked by external circumstances such as attempting to turn midgait or pass through a narrow space such as a doorway. Emotional stimuli including anger or fear can provoke freezing as can the prospect of entering a room filled with people. A variety of sensory or motor tricks such as marching to a cadence are effective in overcoming freezing.
11. Postural reflexes are evaluated by asking the patient to establish a comfortable base, with feet slightly apart and then, while standing behind the patient, applying a brisk backward sternal perturbation. A normal response is to take up to one corrective step backward to prevent falling. When postural reflexes are impaired, more than one step will be needed before balance is reestablished. When postural reflexes are absent, the patient will continue to reel backward and fall if not checked by the examiner.
B. Non-Parkinsonian neurologic signs. Several neurologic findings are associated with one or more forms of atypical parkinsonism, but most of these signs are uncommon in IPD.
1. Apraxia should be tested independently in both upper extremities. The patient should be asked to perform such tasks as saluting, throwing a kiss, or demonstrating how to use an imaginary tooth brush. Inability to perform these tasks in the face of normal strength and coordination, or the use of a body part such as a finger in place of an imagined implement, suggests apraxia. Apraxia and parkinsonism can be seen in cases of corticobasal degeneration (CBD) and AD.
2. Cortical sensory functions such as graphesthesia, stereognosis, and tactile localization are sometimes abnormal in CBD.
3. The alien limb phenomenon is present when a patient manifests uncontrollable grasping and manipulating of objects or when a hand exhibits interfering involuntary movement with one of the other limbs (intermanual conflict). This phenomenon may be present in CBD, ischemic strokes, or Creutzfeldt–Jakob disease (CJD).
4. Ocular motility abnormalities. Inability to generate normal saccadic eye movements, especially downward, with preservation of the same movements when eliciting the oculocephalic reflex, indicates a supranuclear gaze palsy. This finding is most characteristic of PSP but can be found in other forms of atypical parkinsonism as well. It is important to remember that limited upgaze is not an uncommon finding in the normal elderly patient, but impaired downgaze is always abnormal. Excessive macro square wave jerks and spontaneous repetitive small horizontal oscillations of the eyes from the midline are also seen in PSP.
5. Reflex myoclonus, elicited by tapping the arm, leg, or fingertip with the examiner’s own fingertip or with a percussion hammer, may be present in cases of CBD.
6. Blood pressure must be measured in the recumbent and standing positions while recording the concurrent heart rate. Orthostatic hypotension is an early and common manifestation of MSA but occurs later in the course of IPD, especially with the use of dopaminergic or anticholinergic drugs.
7. Mental status evaluation. Evaluation should include functions such as immediate and short-term recall, orientation, constructional praxis, calculation, and comprehension of three-step commands.
8. Other neurologic signs. To determine the full extent of involvement of the central nervous system (CNS), a complete neurologic examination should be performed to establish the presence of hyperactive or hypoactive muscle stretch reflexes, sensory loss, cranial nerve dysfunction, cerebellar signs, pathologic reflexes (especially Babinski’s sign), weakness, or muscle atrophy.
LABORATORY STUDIES
A. Neuroimaging.
1. IPD. In classical IPD where the diagnosis is strongly suggested by the history and physical examination, neuroimaging is not necessary. IPD is commonly asymmetric, but if symptoms or signs of parkinsonism are remarkably asymmetric resulting in severe involvement on one side and virtually no involvement on the other, a CNS imaging study, preferably magnetic resonance imaging (MRI), is indicated to evaluate for the possibility of unilateral structural basal ganglia pathology such as a neoplasm, arteriovenous malformation, infarction, or the presence of brain hemiatrophy.
2. Other forms of parkinsonism. In patients with insufficient findings to make a diagnosis of IPD (e.g., a patient with hypokinesia only) or with additional neurologic findings not usually seen in IPD, a brain imaging procedure is indicated, preferably an MRI. Not all degenerative forms of parkinsonism are associated with demonstrable MRI abnormalities and those that are may demonstrate the characteristic abnormality infrequently or only in the advanced stages of the illness (see Section C under Diagnostic Approach). Therefore, a normal MRI or computed tomography (CT) scan of the brain does not rule out syndromes such as PSP or MSA but does usually rule out normal pressure hydrocephalus (NPH), brain tumor, or stroke.
B. Laboratory and genetic tests are not useful in establishing a diagnosis of IPD except in few genetic forms, but can be of benefit in diagnosing several other causes of parkinsonism (see Section G under Diagnostic Approach).
DIFFERENTIAL DIAGNOSIS
A. IPD. This is the most common cause of parkinsonism with a prevalence 0.2% and increasing with age (4% among those patients older than 80 years). IPD is a degenerative disorder of unknown but probable multifactorial etiology, with genetics likely conferring susceptibility to the effects of the environment and aging in most cases. More than 10 mutations (e.g., Parkin, PINK1, LRRK2) with Mendelian pattern of inheritance have been identified, leading to an IPD picture ranging from young onset with some atypical features to typical presentation and course in old age. Yet, patients with single-gene disorders constitute a small minority of the overall PD population. Consideration of single-gene inheritance is most important in young-onset patients. Metabolic dysfunction of mitochondrial complex I has been demonstrated in PD, whether acquired or hereditary. The predominant abnormality is in the substantia nigra pars compacta and nigrostriatal pathway leading to dopamine deficiency in the striatum. The wide spectrum of symptoms and the resistance of some nonmotor symptoms (such as depression, sleep disorders, cognitive impairment, and autonomic dysfunction) to levodopa support the pathologic observations that the degenerative process also involves other brainstem nuclei and subcortical structures.
1. Clinical. The cardinal symptoms are resting tremor, bradykinesia, rigidity, and impairment of postural reflexes. The onset is usually asymmetric, and tremor is the most common presenting sign. Postural instability, gait difficulty, and dysautonomia appear with progression of the disease. Depending on the age of the cohort and follow-up period, 30% to 78% of patients have been reported to develop dementia, but it is seldom severe and is never a presenting symptom. The incidence of an IPD increases sharply with age, although it can present at any age. Arbitrarily, patients with onset between ages 21 and 39 are classified as young-onset IPD. They exhibit a more gradual progression of symptoms and are more likely to experience dystonia as an early sign. Levodopa-induced dyskinesias and motor fluctuations that can occur in IPD at any age are more frequently observed in this age group. The differential diagnosis of juvenile parkinsonism (before the age of 21) is broad and includes hereditary and metabolic conditions.
2. Neuroimaging. MRI and CT of the brain are usually unremarkable in IPD. A positron-emission tomography (PET) scan shows decreased fluorodopa uptake in the striatum but no striatal abnormality in fluorodeoxyglucose scans. Single photon emission computed tomography (SPECT) shows decreased dopamine transporter density, although it must be kept in mind that some other forms of parkinsonism, such as MSA and PSP, can also result in an abnormal SPECT scan.
3. Neuropathology. Lewy bodies (eosinophilic intra cytoplasmic inclusions), mainly in the substantia nigra, are the pathologic hallmark of this disorder. In IPD, these inclusions stain for alpha-synuclein, the protein produced by the mutant gene in the rare autosomal-dominant form of PD.
4. Other tests. There is no specific test for the diagnosis of IPD.
B. Secondary parkinsonism. Parkinsonism can be induced by a wide spectrum of disease processes affecting the brain, especially the basal ganglia. These include infection, cerebrovascular disorders, toxins, metabolic disorders, trauma, neoplasm, drugs, hypoxemia, and hydrocephalus. Selected causes include the following:
1. Drug-induced parkinsonism. Neuroleptics and metoclopramide block striatal D-2 dopamine receptors, whereas reserpine depletes dopamine from presynaptic vesicles. Each of these drugs can result in motoric symptoms indistinguishable from IPD. The “atypical” neuroleptic clozapine mainly blocks extrastriatal (D-4) receptors and does not cause parkinsonism; quetiapine also seems to have a low potential to cause this adverse effect. Other atypical neuroleptics, such as risperidone, olanzapine, and aripiprazole, can cause parkinsonism. An underlying predisposition to PD may be in part responsible for the emergence of this syndrome. The resolution of drug-induced parkinsonism may take several months after discontinuation of the offending medication.
a. Neuroimaging. SPECT scan of the dopamine transporter protein is very useful in distinguishing drug-induced parkinsonism from IPD in that SPECT is normal in the former condition.
2. Normal pressure hydrocephalus.
a. Clinical. This is a form of communicating hydrocephalus. Approximately one-third of patients with this disorder have a history of spontaneous or traumatic subarachnoid hemorrhage or meningitis. Although, as measured by lumbar puncture, the cerebrospinal fluid (CSF) pressure is normal, there is excessive force on the walls of the dilated lateral ventricles, especially the frontal horn, leading to the compression of surrounding structures. The clinical triad of NPH consists of gait apraxia (magnetic gait), subcortical dementia (which may later include cortical features), and urinary incontinence, often appearing late in the illness. The hesitant gait may resemble that seen in IPD, but the absence of rest tremor, the appearance of incontinence, and the absence of significant benefit from levodopa allow the two conditions to be distinguished. Early recognition of this syndrome is important because in some cases shunting the ventricles can reverse it (see Chapter 8).
b. Neuroimaging. Enlarged lateral ventricles, especially the frontal and lateral horns, which are disproportionate to cortical atrophy, are seen. A proton density MRI demonstrates periventricular hyperintensity suggesting transependymal flow.
c. Other tests. Fisher’s test consists of removing 30 to 50 cc of CSF and observing for improvement in symptoms over the next 24 hours. It is a useful test and does not require sophisticated laboratory techniques. Intracranial pressure monitoring allows demonstration of periods of high CSF pressure (b-waves) and is used widely as a predictor of response to shunting.
3. Hemiatrophy–hemiparkinsonism. These patients present at a relatively early age with markedly asymmetric parkinsonism affecting the side of their body manifesting hemiatrophy. They may have a history of abnormal birth and contralateral hemisphere hemiatrophy, both of which raise the possibility of an early childhood brain insult, which later in life manifests as delayed-onset parkinsonism. The slow progression of this disorder, its occasional association with dystonia, and the striking asymmetry form the basis of its distinction from IPD.
4. Toxins.
a. Carbon monoxide (CO). Acute or chronic CO poisoning causes globus pallidus or striatal necrosis. The onset of parkinsonism can be immediate after the incident, but more commonly develops days to weeks after an initial recovery from coma. Response to levodopa is poor or absent.
b. Manganese intoxication can result in a parkinsonian state, and in addition is often associated with unusual behavioral symptoms such as hallucinations and emotional lability or other movement disorders such as dystonia.
c. Cyanide and methanol intoxication can also cause bilateral basal ganglia necrosis and parkinsonism.
5. Cerebrovascular disease. Either a lacunar state with multiple small infarcts of the basal ganglia or subacute arteriosclerotic encephalopathy affecting basal ganglia connections can lead to parkinsonism. In either condition, dementia is also common. Resting tremor is usually absent in these patients. Gait disorder can be very prominent and occasionally constitutes the only neurologic symptom, giving rise to the term “lower-body parkinsonism.” The response to levodopa is limited, but occasional patients do show benefit.
10. Trauma. Pugilistic encephalopathy is a progressive neurologic syndrome characterized by parkinsonism, dementia, and ataxia. It is seen in boxers with a history of repeated head trauma. Treatment is usually unsatisfactory.
Focal acute injury to the midbrain and substantia nigra and subdural hematoma are two other possible causes of posttraumatic parkinsonism.
C. Parkinson-plus syndromes. This is a group of parkinsonian syndromes distinguished from IPD by the presence of additional prominent neurologic abnormalities. In these conditions, there may be cerebellar, autonomic, pyramidal, oculomotor, cortical sensory, bulbar, cognitive, and psychiatric dysfunction, as well as apraxia and movement disorders not typically seen in untreated IPD such as myoclonus, dystonia, or chorea. Any of these neurologic or psychiatric abnormalities can appear early in the course of the illness. Early falls with gait disturbance or postural instability, absence of resting tremor, early dementia, and supranuclear gaze palsy are signs that should always prompt consideration of a Parkinson-plus syndrome. The parkinsonian components of these disorders such as akinesia and rigidity are usually not responsive to levodopa, although early transient responsiveness can be observed. The onset of these diseases is generally in the fifth or sixth decade of life with average survival of 5 to 10 years. The cause of death is usually pneumonia, other intercurrent infections, or sepsis. The etiology of this entire group of disorders is largely unknown.
Despite the apparent clinical differences between IPD and the Parkinson-plus syndromes, differentiation between the two can be difficult. In a clinicopathologic study, 24% of patients who were clinically diagnosed with IPD were found to have a different type of parkinsonism at autopsy. In Parkinson-plus syndromes, the brain MRI can occasionally be helpful. An electroencephalogram (EEG) may show nonspecific abnormalities such as slowing of the background activity. The specific clinical and imaging features of individual Parkinson-plus syndromes are described below. Although each of these conditions has characteristic clinical findings, it is important to remember that there is significant overlap in signs and symptoms among them.
1. PSP.
a. Clinical. Early onset of gait difficulty, loss of postural reflexes resulting in backward falls, and freezing of gait, coupled with supranuclear gaze palsy (initially downgaze), are suggestive of PSP. Axial rigidity and nuchal dystonia with extensor posture of the neck, generalized bradykinesia, “apraxia” of eyelid opening and closing, blepharospasm, a furrowed forehead leading to a fixed facial expression, and a monotonous, but not hypophonic voice are additional features suggesting the diagnosis. There is variable, but often mild, cognitive decline, especially in executive functions. The presence of prominent bradykinesia in association with the fixed facial expression raises the possible diagnosis of IPD in these patients, but the ocular motility abnormalities, the early gait instability, the frequent absence of tremor, and the absence or loss of levodopa response suggest the correct diagnosis.
b. Neuroimaging. Midbrain and, later, pontine atrophy are sometimes apparent on MRI.
c. Neuropathology. PSP is a tauopathy. Globose neurofibrillary tangles composed of tau filaments are present affecting mainly the cholinergic neurons of the basal ganglia and brainstem nuclei with apparent sparing of the cortex.
2. CBD.
a. Clinical. More recently, this has often been referred to as cortical basal syndrome, to emphasize the point that different pathologies, most notable PSP, can result in a nearly identical clinical syndrome, which can only be determined at autopsy. This syndrome can present as a strikingly asymmetric or unilateral akinetic-rigid syndrome associated with limb apraxia, alien limb phenomenon, cortical sensory signs, stimulus sensitive myoclonus, dystonia, and postural or action tremor. Supranuclear gaze palsy, cognitive impairment, and pyramidal tract signs can also be seen.
b. Neuroimaging. MRI or CT of the brain is abnormal in some patients and reveals asymmetric frontoparietal atrophy.
c. Neuropathology. CBD is also a tauopathy. Neuronal loss and gliosis are found in the frontoparietal regions and substantia nigra pars compacta. Swollen achromatic neurons and basophilic nigral inclusions, which represent an overlap with Pick’s disease, are characteristic. Abundant cytoplasmic inclusions consisting of aggregated hyperphosphorylated tau protein are found.
3. MSA.
a. Clinical. Sporadic, progressive disease in adults (onset after 30 years of age) characterized by autonomic failure, including urinary incontinence (with erectile dysfunction in men), or an orthostatic decrease in blood pressure by at least 30 mm Hg systolic or 15 mm Hg diastolic within 3 minutes of standing, plus one of the following:
(1) Parkinsonism (slowness of movements, rigidity, and tendency to fall) with poor response to levodopa (parkinsonian subtype [MSA-P]). MSA-P was referred to as striatonigral degeneration before.
(2) A cerebellar syndrome (wide-based gait, uncoordinated limb movements, action tremor, and nystagmus) (cerebellar subtype [MSA-C]). MSA-C was referred to as olivopontocerebellar atrophy before.
From a diagnostic point of view, MSA should always be suspected in the hypokinetic patient with little response to levodopa who also manifests prominent autonomic or cerebellar dysfunction.
b. Neuroimaging. MRI of the brain shows putaminal hypointensity in MSA-P, probably because of excessive iron deposition in this structure. Cerebellar atrophy can be seen in MSA-C.
c. Neuropathology. Common to all the MSA syndromes is the presence of characteristic glial cytoplasmic inclusions. Like Lewy bodies seen in IPD, these inclusions stain for the protein alpha-synuclein. Especially in SDS, additional neuronal loss and gliosis are seen in the structures responsible for autonomic functions such as the intermediolateral cell column of the spinal cord and the dorsal motor nucleus of the vagus.
(1) Dementia syndromes. AD, Pick’s disease, and DLB are degenerative CNS disorders whose predominant manifestation is dementia. Familial frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) are associated with mutations in the tau gene. Although the degenerative process in these disorders has a predilection for certain cortical regions, subcortical structures may also be involved leading to extrapyramidal manifestations including parkinsonism. The key to identifying a primary dementing disorder as a cause of parkinsonism is the early appearance of dementia, often antedating the onset of parkinsonism (e.g., dementia occurs before or within 1 year after the onset of parkinsonism in DLB).
D. Heredodegenerative diseases.
1. Wilson’s disease. An autosomal-recessive condition associated with impairment of copper excretion caused by a genetic defect in a copper-transporting ATPase, resulting in copper accumulation in different organ systems including the CNS, liver (cirrhosis), cornea (Kayser–Fleischer ring), heart, and kidney.
a. Clinical. The age of presentation ranges from 5 to 50, peaking between 8 and 16. Neurologic symptoms are present at the onset of the disease in about 40% of patients. Extrapyramidal symptoms such as dystonia, rigidity, and bradykinesia are more common in children, whereas tremor and dysarthria are more likely to appear in adults. A variety of psychiatric symptoms can be seen in Wilson’s disease. An especially important clue to the diagnosis is the presence of liver dysfunction such as cirrhosis or chronic active hepatitis, especially in a young patient. The combination of bradykinesia and tremor in these patients may suggest PD, but the very young age of onset, and the presence of psychiatric symptoms, liver dysfunction, or dystonia should prompt a search for laboratory signs of Wilson’s disease. As the consequences of Wilson’s disease are preventable and the neurologic symptoms are reversible with early treatment using copper chelating drugs, this condition should always be kept in the differential diagnosis of atypical parkinsonism, especially that appearing below the age of 50.
b. Neuroimaging. MRI of the brain shows ventricular dilation as well as cortical and brainstem atrophy. The basal ganglia, especially the putamen, may appear either hypo- or hyperintense on T2-weighted studies, and hypodense on CT examinations. Occasionally, there is the characteristic “face of the giant panda” appearance of the midbrain on MRI studies.
c. Neuropathology. There is generalized brain atrophy. The putamen, globus pallidus, and caudate nucleus are cavitated and display a brownish pigmentation reflecting copper deposition.
d. Other tests. Plasma ceruloplasmin is the most useful screening test and is usually below 20 mg/dL (normal: 25 to 45 mg/dL). Plasma copper is decreased and 24-hour urinary copper excretion is increased. Slit lamp examination of the cornea reveals Kayser–Fleischer ring in almost all neurologically symptomatic patients and represents a very specific but not pathognomonic finding. If one or more of these tests are normal and the diagnosis is in doubt, it should be confirmed by liver biopsy that shows increased copper content. Because of abundance of disease-specific mutations and their location at multiple sites across the genome, genetic diagnosis is limited to kindreds of known patients.
2. HD. A relentlessly progressive autosomal-dominant disorder characterized by dementia, psychiatric disturbance, and a variety of movement disorders.
a. Clinical. The major clinical components of HD are cognitive decline, various psychiatric abnormalities (personality changes, depression, mania, and psychosis), and movement disorder. Although chorea is the most common motoric symptom, bradykinesia usually coexists with chorea and may explain the occasional exacerbation of the motor impairment when control of the chorea is attempted with antidopaminergic medications. An abnormality of saccadic eye movement, particularly slow saccades, is often one of the earliest neurologic signs of this disorder. The typical age of onset is in the fourth or fifth decade, but 10% of the patients develop symptoms before age 20 (juvenile Huntington’s). Successive generations may develop symptoms at a progressively earlier age, especially if they have inherited the disease from their father, reflecting the genetic phenomenon of anticipation. The juvenile form presents with a combination of a progressive akinetic-rigid syndrome (Westphal’s variant), dementia, ataxia, and seizures. It is these akinetic-rigid patients that are most likely to be confused with IPD, but the autosomal-dominant inheritance pattern, the early age of onset, and the presence of seizures should suggest the correct diagnosis. The duration of illness from onset to death is about 15 years for adult-onset HD and 8 to 10 years for those with onset in childhood.
b. Neuroimaging. Atrophy of the head of the caudate is the principal finding on neuroimaging. It can be appreciated on either MRI or CT scan.
c. Neuropathology. There is loss of medium spiny striatal neurons, as well as gliosis in cortex and striatum (particularly the caudate). This striatal neuronal loss accounts for the drastic decrease in the two neurotransmitters associated with these cells, -aminobutyric acid, and enkephalin.
d. Other tests. HD can be diagnosed and presymptomatic individuals can be identified with great certainty using DNA testing. The genetic abnormality has been localized to chromosome 4 and consists of an expansion of the usual number of repeats of the trinucleotide sequence CAG. The presence of 40 or more CAG repeats confirms the diagnosis of HD. Reduced penetrance is seen with 36 to 39 CAG repeats. Because of the ethical, legal, and psychological implications of presymptomatic predictive testing, it should only be carried out by a team of clinicians and geneticists fully sensitive to these issues and aware of published guidelines.
3. Other neurologic conditions, occasionally associated with parkinsonism, include neuroacanthocytosis, neurodegeneration with brain iron accumulation (NBIA, formerly known as Hallervorden–Spatz syndrome), Machado–Joseph disease (spinocerebellar ataxia type 3), Fragile X-associated tremor/ataxia syndrome (FXTAS), and familial calcification of the basal ganglia.
DIAGNOSTIC APPROACH
A. Clinical. Careful history taking and physical examination are essential. A meticulous survey of the past medical and psychiatric history, family history, and occupational or environmental exposure to toxins will reveal most causes of secondary parkinsonism. Disease onset at a young age, a strong family history of the same disorder, lack of resting tremor, absent response to levodopa and early appearance of postural instability, gait disorder, dysautonomia, or dementia should be considered red flags in the history suggesting a diagnosis other than IPD. The general physical examination is important because it may reveal signs of a systemic disease that is contributing to secondary parkinsonism. Neurologic examination establishes whether parkinsonism is isolated or associated with involvement of other neuronal systems in the CNS. The presence of aphasia, apraxia, supranuclear gaze palsy, cortical sensory loss, alien limb phenomenon, pyramidal signs, lower-motor neuron findings, myoclonus, chorea, or dystonia indicates more widespread CNS involvement than is the case in IPD.
B. General laboratory tests.
1. CBC and peripheral blood smear. Acanthocytes are found on a fresh peripheral blood smear in neuroacanthocytosis. A low hemoglobin level and elevated reticulocyte count consistent with hemolytic anemia may be present in Wilson’s disease.
2. Blood chemistry. Abnormal liver function tests may be found in Wilson’s disease. Hypocalcemia, hypomagnesemia, and a low parathormone level are present in hypoparathyroidism. Elevated creatine kinase is associated with neuroacanthocytosis, and elevated serum lactate, suggesting lactic acidosis, is found in mitochondrial cytopathies. Low thyroxin and high thyroid-stimulating hormone (TSH) levels point to hypothyroidism.
3. Serology. Elevated ESR, C-reactive protein, or rheumatoid factor may be found in inflammatory or rheumatologic conditions. Antibodies against glutamic acid decarboxylase are present in stiff person syndrome.
C. Radiology.
1. Plain X-rays. Spine X-rays may reveal ankylosing spondylitis or osteoarthritis as the cause of mechanical limitation of movement.
2. CT or MRI of the brain. CT may demonstrate a neoplasm, stroke, hydrocephalus, basal ganglia calcification, atrophy, or sequelae of trauma. It has some limitations, in that the resolution is not always adequate to evaluate density changes or storage materials in the basal ganglia, and brainstem or cerebellar cuts may suffer from bone artifact (see Chapter 35). In these circumstances, an MRI of the brain is more desirable. Several characteristic MRI patterns that are suggestive of specific hypokinetic disorders are listed below:
a. Many lacunar strokes. vascular parkinsonism
b. Large ventricles, out of proportion to cerebral atrophy; transependymal flow: NPH
c. Caudate atrophy. HD
d. Decreased T2 signal in striatum. MSA
e. Homogeneous decreased T2 signal or decreased T2 signal with a central hyperintensity (Tiger’s eye) in the globus pallidus: NBIA
f. Striatal necrosis. Wilson’s disease, Leigh’s disease, and CO intoxication
g. Midbrain atrophy. PSP
h. Asymmetric frontoparietal atrophy. CBGD
3. PET or SPECT. With modern analysis techniques, fluorodeoxyglucose PET, by characterizing the regional cerebral metabolism pattern, can distinguish PD, MSA, and PSP from one another with >90% accuracy. These techniques are not readily available at many hospitals, however. The status of nigral dopaminergic neurons can be determined using fluorodopa PET or [I123]FP-CIT (Ioflupane) SPECT. In IPD, either of these two modalities demonstrates a loss of dopaminergic nigral cells. Although both of these techniques identify nigral dopaminergic dysfunction, they do not clearly differentiate between IPD and other causes of parkinsonism such as MSA, PSP, and CBD. The major clinical usefulness of Ioflupane SPECT is that it is very accurate in distinguishing IPD from mimicking conditions that do not involve dopamine-producing cells such as ET, dystonic tremor, or drug-induced parkinsonism. Of additional importance, SPECT imaging equipment is available at many hospitals.
D. Electrophysiology.
1. ECG. Heart block may be present in mitochondrial cytopathy.
2. EEG. Epileptic activity or focal slowing may appear with focal lesions (stroke and tumor). Slow background activity is seen in some primary dementias. Periodic triphasic complexes may be present in CJD (see Chapter 36).
3. EMG/nerve conduction studies. Mild nerve conduction slowing suggestive of axonal polyneuropathy is seen in neuroacanthocytosis. Myopathic findings on EMG (see Chapter 36) may be present in cases of mitochondrial cytopathies.
E. Neuropsychological testing. If there is clinical suspicion of dementia, formal testing should be employed to plot the profile of cognitive decline (see Chapter 4).
F. CSF analysis. Elevated protein and pleocytosis can be detected in CNS infections. The presence of high levels of the 14-3-3 protein in CSF is highly suggestive of CJD (see Chapter 36). A large volume of CSF can be removed (Fisher’s test) with observation for improvement in neurologic signs as one means of corroborating the diagnosis of NPH.
G. Special diagnostic tests.
1. Wilson’s disease. Low ceruloplasmin, low serum copper, increased 24-hour urinary copper excretion, and Kayser–Fleischer ring on slit lamp examination of the cornea are all suggestive of Wilson’s disease. Liver biopsy for copper content is performed only if the diagnosis is in question.
2. NPH. Intracranial pressure monitoring shows episodic appearance of high-pressure waves.
H. Genetic testing. Monogenic PD is found in approximately 3% of IPD patients and mutations in these PD genes are most common in those with an early age of onset or those belonging to certain ethnic groups. Commercial testing is available for LRRK2, PINK1, DJ-1, SNCA (alpha-synuclein), GBA (glucocerebrosidase), and Parkin genes. In patients with onset before age 51, almost 20% have a mutation in one of these genes, most commonly Parkin, followed by LRRK2. The mutation rate is still higher for those with onset prior to age 30. In individuals developing PD under the age of 20, as many as 77% have a mutation of the Parkin gene. Jewish PD patients are more likely to harbor a mutation of the GBA gene. Although genetic testing does not affect patient management, it can clarify prognosis and allow genetic counseling. Genetic testing is also increasingly more available for other conditions where parkinsonism can be a clinical component of the overall syndrome such as HD, FXTAS, SCA3, and other SCA subtypes, dystonia subtypes, and various mitochondrial conditions.
I. Therapeutic trial. A brisk and unequivocal beneficial response to a trial of levodopa therapy is strongly suggestive of IPD. It must be kept in mind, however, that in some of the Parkinson-plus syndromes there can be a positive response, although seldom remarkable, frequently only present with large doses, and often not persistent over time.
WHEN TO REFER
Patients with new-onset hypokinesia who have the following characteristics are less likely to have IPD and would benefit from referral to a movement disorders specialist:
A. Early onset, for example, before 50 years of age
B. Early gait difficulty and postural instability
C. Prominent dementia
D. A family history of parkinsonism
E. Supranuclear gaze palsy
F. Apraxia, alien limb phenomenon, cortical sensory loss, myoclonus, marked asymmetry of neurologic involvement
G. Bulbar, cerebellar, or pyramidal dysfunction
H. Marked dysautonomia
I. Absent, limited, or unsustained response to levodopa.
Key Points
• IPD is the most common cause of hypokinesia.
• Aside from IPD, the differential diagnosis of hypokinesia includes the Parkinson-plus syndromes, NPH, fragile X tremor ataxia syndrome, some spinocerebellar ataxia syndromes, Wilson’s disease, and (especially juvenile) Huntington’s disease.
• In addition to hypokinesia, IPD is characterized by rigidity, rest tremor, loss of postural reflexes, and a great number of nonmotor features including, but not limited to, autonomic dysfunction, depression, dementia, and sleep disorders.
• SPECT imaging of the dopamine transporter is very useful in establishing a diagnosis of non–drug-induced parkinsonism, but not in distinguishing IPD from Parkinson-plus syndromes.
• Genetic analysis of patients with hypokinesia is becoming increasingly important and more available in the case of IPD, especially in those patients with a very young age at onset or a strong family history. It is extremely important for the evaluation of spinocerebellar ataxia syndromes, Huntington’s disease, dystonia, and fragile X tremor ataxia syndrome.
• An unequivocal and lasting response to levodopa therapy is generally an important feature that distinguishes IPD from most other conditions presenting with hypokinesia.
Related posts:
 Approach to the Patient with Acute Confusional State (Delirium/ Encephalopathy)
Approach to the Patient with Sleep Disorders
Approach to the Patient with Hearing Loss
Approach to the Patient with Lower Extremity Pain, Paresthesias, and Entrapment Neuropathies
Approach to Common Office Problems of Pediatric Neurology
Movement Disorders
Approach to the Patient with Acute Confusional State (Delirium/ Encephalopathy)
Approach to the Patient with Sleep Disorders
Approach to the Patient with Hearing Loss
Approach to the Patient with Lower Extremity Pain, Paresthesias, and Entrapment Neuropathies
Approach to Common Office Problems of Pediatric Neurology
Movement Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


