Introduction
Most of our views concerning the fundamental neuronal mechanisms of epileptic phenomena derive from investigations carried out on experimental animal models. An anonymous bioscientist, however, once stated, “The best model of a cat is a cat, and preferably the same cat,” implying that the ideal approach to studying basic mechanisms of human epilepsy is actually to study patients with epilepsy. Although direct examination of the epileptic human brain has become much more feasible in recent years, experimental paradigms are severely limited by cost and ethical considerations. As a result, animal models remain an essential part of basic research into the pathophysiology of human epilepsy. Animal models are invaluable for expanding upon concepts, derived from observations of patients, that cannot be pursued with further human experimentation. Conversely, research on patients with epilepsy is essential to validate the relevance of data obtained from animal models. Both these goals are best met by designing parallel investigations involving patients with epilepsy and experimental animal preparations. Such parallel studies are greatly facilitated today by new clinical tools, such as advanced noninvasive structural and functional neuroimaging, and the unique opportunities for invasive investigations in vivo and the utilization of human tissue in vitro that exist in association with the surgical treatment of epilepsy.38,48,102 Progress in molecular genetics promises yet another powerful means, not only for establishing similarities between specific animal models and human epileptic disorders, but also for defining epileptogenic defects in the human brain that can be precisely replicated in the animal laboratory.
There are many forms of human epilepsy, which undoubtedly reflect a variety of pathophysiologic mechanisms. In fact, a single, well-defined human epileptic condition, and even an individual epileptic seizure, involves several specific disturbances in neuronal function that occur simultaneously or in sequence before clinical manifestations appear. To carry out parallel studies in patients and animals, it is important to realize that few, if any, human epileptic conditions can be faithfully modeled in toto in the experimental animal laboratory. Consequently, it is necessary to identify and define component parts of human epilepsy that can be more easily investigated individually and replicated in animals.33,35 This chapter, therefore, first considers one strategy for dissecting out components of epilepsy, and then examines what has been learned about the basic mechanisms of these component parts from parallel studies of the human condition and appropriate animal models.
Components of Human Epilepsy
An epileptic seizure is an acute event that can be a natural, albeit pathologic, reaction of a normal brain to a noxious insult. Although an epileptic seizure that has been acutely induced in an animal model may replicate certain aspects of epileptic seizures occurring in patients with epilepsy, the underlying intrinsic and enduring epileptogenic abnormality responsible for recurrent epileptic seizures in patients with epilepsy is missing in this situation. Consequently, investigations of acute seizures induced in otherwise normal experimental animal models provide limited information about the pathophysiologic substrates responsible for repeated spontaneous seizure generation in patients. Nevertheless, much information can be gained about mechanisms of ictal precipitation, ictal propagation, ictal termination, and postictal phenomena.
Epileptogenesis, or the mechanism by which an epileptic condition is acquired, undoubtedly takes many forms, but it is most evident in secondary (symptomatic) human epileptic conditions, in which habitual recurrent seizures begin many months or even years after lesions occur in the brain. Epileptogenesis has been studied in animals by introducing into the brain certain metals, such as alumina cream, and more recently by inducing status epilepticus (SE) with excitotoxins, such as kainic acid (KA) and pilocarpine, or electrical stimulation; this results in structural lesions and the development of recurrent spontaneous seizures.95 The process of epileptogenesis has also been brought under more precise laboratory control with the various kindling models of epilepsy.80
Just as basic research on epilepsy made a paradigm shift with the realization that animals with chronic epilepsy more faithfully model the human condition than do epileptic seizures induced in a normal brain, investigations with animal models now requires a second paradigm shift. Given that a genetic predisposition often exists to symptomatic human epilepsies, identification of susceptibility genes and other predisposing factors should make it possible to create more realistic animal models of epileptogenesis by inducing a chronic epileptogenic lesion in a brain with these predisposing factors.40
The interictal state represents a period of relative quiescence, during which the enduring pathophysiologic disturbances that predispose to the spontaneous recurrence of epileptic seizures are minimally active, or held in abeyance by some active seizure-suppressing mechanism. These diverse phenomena can be studied in the laboratory, using animal models of chronic epilepsy in which seizures are generated spontaneously.
Ictal onset represents a functional transition from the interictal state; it can reflect a variety of processes, depending on the underlying pathophysiology and the type of ictal manifestation.34 Increases in excitation or synchrony, decreases in seizure-suppressing mechanisms, and nonspecific precipitating factors can be studied during transition to ictus in animal models of chronic epilepsy.
Many types of seizures occur, and the ictal state undoubtedly reflects a variety of aberrant neuronal mechanisms involving different patterns of excitatory and inhibitory events, different synchronization processes, and different brain regions and pathways. Individual ictal events often also show evolution,
involving a variety of forms of propagation of ictal discharge and resulting in variable ictal mechanisms specific to the region to which propagation has occurred. With focal seizures, this includes local spread, propagation to structures that impair consciousness when simple partial seizures evolve to complex partial seizures, and propagation to structures that generate generalized convulsive manifestations when seizures secondarily generalize. Such propagation patterns, and peculiarities of ictal discharges in particular brain regions, can be studied in the animal laboratory with acute as well as chronic models. However, pathways, and therefore ictal spread, vary across species and may not accurately replicate the human condition; for example, interhippocampal connections are strong in rats and weak in humans, a difference that can affect the manifestations of temporal lobe seizures.121
involving a variety of forms of propagation of ictal discharge and resulting in variable ictal mechanisms specific to the region to which propagation has occurred. With focal seizures, this includes local spread, propagation to structures that impair consciousness when simple partial seizures evolve to complex partial seizures, and propagation to structures that generate generalized convulsive manifestations when seizures secondarily generalize. Such propagation patterns, and peculiarities of ictal discharges in particular brain regions, can be studied in the animal laboratory with acute as well as chronic models. However, pathways, and therefore ictal spread, vary across species and may not accurately replicate the human condition; for example, interhippocampal connections are strong in rats and weak in humans, a difference that can affect the manifestations of temporal lobe seizures.121
Ictal termination also takes many forms. It undoubtedly involves both active and passive neuronal mechanisms, which vary according to seizure type and underlying pathophysiology. Even though ictal termination can be easily investigated using animal models of acute seizures, relatively little research has been carried out on this phenomenon, compared with the attention given to ictal onset.25
The postictal period can be associated with brief or prolonged neuronal dysfunction, ranging from isolated focal neurologic deficits to coma. These clinical disturbances presumably reflect, at least in part, the enduring effects of active mechanisms that act to terminate the ictal state.16
Interictal behavioral disturbances can represent the enduring consequences of repeated epileptic seizures or can be produced by the underlying brain disturbance/damage that is responsible for the seizures themselves. Underlying mechanisms that have been implicated in long-term effects of epilepsy include (a) specific structural damage, which might be the result, rather than the cause, of seizures; (b) transsynaptic functional changes, such as those modeled by kindling; and (c) side effects of natural seizure-suppressing mechanisms that are necessary to maintain the interictal state. Some animal research is being carried out to elucidate mechanisms of epilepsy induced interictal behavioral disturbances. However, given the degree of clinical disability that can be attributed to these disturbances, more effort in this area of research should be encouraged.39
Studies of Human Epilepsy
With the preceding dissection of epileptic phenomena as a framework for organizing the approaches to research into basic mechanisms of human epilepsy, the remainder of this chapter focuses on how the results of investigations of human epilepsy confirm or contradict concepts derived from the results of studies carried out on experimental animal models.
Epileptogenesis and the Interictal State
The primary (idiopathic) epilepsies are genetically transmitted, typically have an age-related onset, and may resolve spontaneously after several years. These disorders are presumably caused by defects that manifest themselves as epileptic seizures only at vulnerable periods during cerebral development. Epileptogenesis, or the active process of creating an epileptic condition, is therefore difficult to evaluate in these conditions—although it is certainly possible that genetically determined abnormalities may be elaborated as the brain matures, and thus a process of epileptogenesis is likely to also occur in these types of epilepsies. The secondary (symptomatic) epilepsies, however, usually result from structural disturbances that become epileptogenic lesions during some finite period. In patients with epilepsy caused by metabolic diseases, congenital malformations, or intrinsic substrates such as neoplasia, the timing of this process is undeterminable; however, in others (e.g., those in whom epilepsy develops following head trauma or a cerebrovascular accident), the existence of a well-defined “latent period” is well documented. Presumably, the damaged brain reorganizes during this period in a manner that ultimately predisposes to the recurrence of detectable, spontaneous epileptic seizures.
Clinical data indicate that the likelihood that epilepsy will develop after an epileptogenic insult depends on the area of brain damaged, type of damage, age at which the damage occurred, and genetic predisposition.70 It is clear from clinical experience that mechanisms accounting for the occurrence of acute seizures at the time of, or shortly after, an epileptogenic cerebral insult are different from those accounting for the development of a chronic epileptic condition many months or years later; that is, early seizures do not necessarily predict late seizures, and the risk factors for epileptic seizures occurring at the time of head trauma or stroke are not exactly the same as those for the development of late posttraumatic epilepsy70 (Table 1), although early seizures may indicate an increased risk of later development of epilepsy.70 Furthermore, whereas the immature brain is more susceptible to acute seizures than the mature brain, it is less likely than the mature brain to generate subsequent spontaneous recurrent seizures, given the same initial insult.91,101 The clinical expression of the process of epileptogenesis in the human brain (e.g., clinical seizures) following injury-associated insult can require considerable time, although this is not well understood.55 The anatomic reorganizations that are currently hypothesized to underlie the epileptogenic process, however, cannot be the only epileptogenic factors involved, because the latent period can be much longer than expected for such changes to take place; the increased risk for epilepsy after head trauma persists for well over 10 years,70 even though chronic epilepsy develops in more than half of such cases within the first year.
Table 1 Risk factors for epileptic seizures and epilepsy after nonmissile head trauma | ||
|---|---|---|
|
It is also important to note that the brain reorganization underlying epileptogenesis (see the next section) is not necessarily a product of an epileptogenic insult, but may be determined by genetic factors (i.e., aberrant brain developmental processes). At the very least, it is quite clear from studies of animal models that genetics contribute significantly to epilepsy predisposition.118
Our current understanding of the basic mechanisms of human epileptogenesis derives largely from studies of patients with mesial temporal lobe epilepsy (TLE), perhaps the most common human epileptic condition.103 Because mesial TLE is often medically refractory and treated preferentially by surgical resection, extensive diagnostic tests—at times involving in vivo invasive electrophysiologic recordings—are carried out, and excised tissue becomes available for in vitro investigations. The pathologic substrate that is most often associated with the clinical condition known as mesial TLE is hippocampal sclerosis.6,23 Microscopic examination of mesial temporal structures has further defined the kinds of lesions seen in the parts of medial temporal lobe removed from patients with hippocampal sclerosis. These include losses of neurons in almost all hippocampal regions, with a heavier loss in the hilus, CA1, and CA3, and lesser losses in CA2 and dentate gyrus (DG) granule cell (GC) layer. In addition, clear synaptic reorganization is present, with collaterals of surviving DG granule cells reinnervating the inner molecular layer of DG GC dendrites, as well as inhibitory interneurons scattered throughout the DG (see Chapter 13). It is becoming increasingly apparent that patients with this condition also have much more widespread extrahippocampal pathological changes as well.120
The lesions seen in mesial TLE with hippocampal sclerosis in human brain have been modeled in the animal laboratory using a variety of techniques that cause hippocampal cell loss and neuronal reorganization.9,75,111,113,115 Whereas the progression of epileptogenic changes can be examined in these animal models, studies of patients with medically refractory mesial TLE reveal only the end results of the epileptogenic process. The pathologic changes observed in these patients could be either the cause or the effect of recurrent epileptic seizures; if the latter, they could result in further epileptogenicity or reflect natural homeostatic mechanisms acting to suppress seizure occurrence. It is also important to note that the sclerotic hippocampus, despite all our attention, may not be the primary epileptogenic region in some patients with mesial TLE. Research results are confounded by the fact that there is no adequate control tissue for human studies, because epileptiform abnormalities can be widespread and often involve both temporal lobes; in addition, evaluations are complicated by the superimposed effects of antiepileptic drugs (AEDs), which have invariably been used.38 Nevertheless, important insights have been derived from anatomic, neurochemical, electrophysiologic, and molecular investigations of this human condition.
Hippocampal sclerosis, induced in rats by intrahippocampal injection of KA, is characterized by general cell loss but more pronounced loss of specific neurons, relative preservation of some inhibitory interneurons, and axonal sprouting resulting in the creation of aberrant circuits, gliosis, neurogenesis, and laminar dispersions of the dentate gyrus.9,64,78,92 All these pathologic changes have been demonstrated in surgically resected specimens of human hippocampal sclerosis, suggesting that the epileptogenic process in human mesial temporal lobe epilepsy resembles that of rats injected with KA.5,23,67,110 In addition, amygdala sclerosis has been described,65 and some animal models show evidence for selective entorhinal cell loss.26 Inflammatory processes also play a role in epileptogenesis in some experimental models.116 Mossy fiber sprouting could create powerful recurrent excitatory feedback that might contribute to the development of epileptiform potentials. Such an enhanced excitatory drive is suggested by the observation of increased extracellular levels of excitatory amino acids obtained during microdialysis of human epileptic brain29 and chronic alteration in glutamate receptors in human epileptogenic tissue.52,53,63 Conversely, there is also evidence that a significant number of aberrant mossy fibers terminate on interneurons, which increases inhibition.106
Investigations of human mesial TLE have yielded inconsistent results concerning inhibitory influences. Inhibitory interneurons appear to be preferentially preserved, and sprouting of inhibitory terminals may also occur.8,22 The results of microdialysis studies are equivocal, with evidence for both increased and decreased release of γ-aminobutyric acid (GABA).27 Positron emission tomography (PET) consistently reveals a reduction in benzodiazepine receptor binding in human epileptogenic regions;60,100 this may merely reflect cell loss, but it could also indicate a selective decrease in postsynaptic inhibitory mechanisms or a receptor downregulation resulting from enhanced presynaptic inhibitory neurotransmitter release.
In vitro electrophysiologic studies of human sclerotic hippocampal slices have been difficult to perform, but there is some suggestive evidence of increased membrane excitability,78 which may be mediated by N-methyl-D-aspartate (NMDA) receptors;66 parallel animal studies also show enhanced GABA-mediated inhibition,90 and some studies on human tissue support this.5,50 In vivo electrophysiologic studies also indicate enhanced inhibitory influences in some areas of the human epileptogenic hippocampus and enhanced excitatory influences in others.19,47,122 Enhanced inhibition of the epileptic hippocampus could reflect natural protective mechanisms; however, it is intriguing to note that enhanced inhibition could also predispose to the appearance of epileptiform hypersynchronization.36 Recordings from resected human tissue in a slice chamber have been somewhat disappointing with regard to recreating the epileptiform discharges often recorded in vivo from the same tissue. Spontaneous interictal discharges are usually lacking, and seizure-like discharges can only be elicited after threshold-lowering maneuvers such as reducing inhibition or raising extracellular potassium (K).14 More recent work suggests that recordings from parahippocampal regions (subiculum and entorhinal cortex) may more closely mimic in vivo phenomena. It has also been suggested that the enhanced excitability and increased synchrony in these regions may be related to altered intracellular chloride concentrations, such that normally inhibitory synaptic actions may become excitatory.18
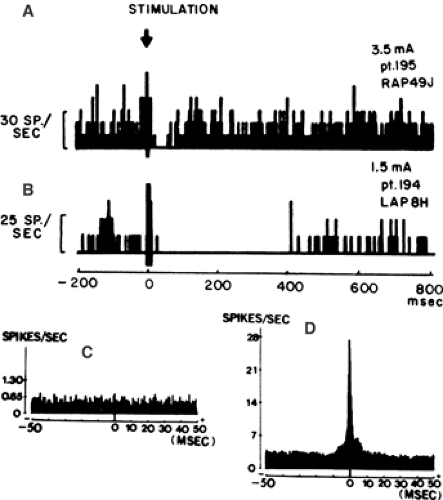 FIGURE 1. Unit histograms from human epileptic hippocampus show weak firing suppression (A) in neurons that were not firing synchronously as determined by cross-correlation histograms (C), and strong firing suppression (B) in neurons that were firing synchronously (D). This provides indirect evidence for recurrent inhibitory circuits as a mechanism of hypersynchronization. From Isokawa-Akesson M, Wilson CL, Babb TL. Prolonged inhibition in synchronously firing human hippocampal neurons. Epilepsy Res. 1989;3:236–247, with permission. |
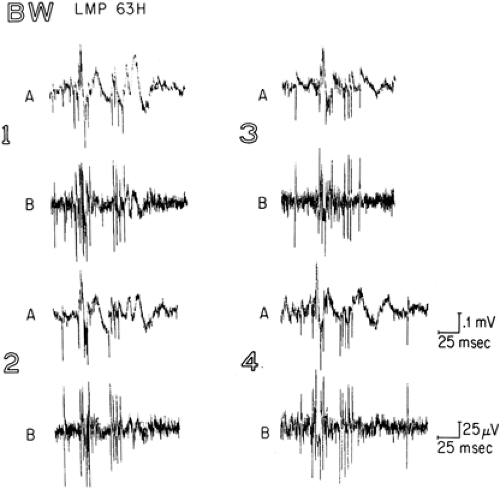 FIGURE 2. EEG spikes (A) and action potentials (B) recorded from the same microelectrode (63H) in the left midhippocampal pes (LMP) in a patient with clinical seizures originating in the LMP. Tracings 1 through 4 were taken from the beginning to the end of the recording period and selected to have a similar EEG spike morphology, characterized by a fast rise time of the initial negative component. Tracing A is a wide-band recording to show the EEG spike-and-slow wave superimposed on the unit discharges. For tracing B, additional high-pass filtering was used to enhance the neuronal discharges. Note increased unit firing during EEG spike, and cessation of unit firing during EEG slow wave. From Babb TL, Crandall PH. Epileptogenesis of human limbic neurons in psychomotor epileptics. Electroencephalogr Clin Neurophysiol. 1976;40:225–243, with permission. |
Unit recordings obtained from human epileptic hippocampus in vivo demonstrate that cells that receive strong inhibitory input are highly synchronized68 (Fig. 1), suggesting that inhibition does play a role in the development of epileptiform hypersynchronization in TLE, just as it does in absence epilepsy83 and in the promotion of normal rhythmic neuronal activity.2
The interictal state in human mesial TLE is characterized by the appearance of interictal electroencephalographic (EEG) spike-and-slow-wave discharges in the epileptogenic temporal lobe, and often independently in the contralateral temporal lobe as well. Microelectrode recordings demonstrate that the spike is associated with increased unit firing, and the slow wave with absence of unit firing (Fig. 2),7 suggesting that this epileptiform transient reflects synchronous paroxysmal depolarization shifts and afterhyperpolarizations of neurons within the epileptogenic hippocampus, in much the same way as occurs in the experimental penicillin focus.24 These abnormal excitatory and inhibitory events occur relatively infrequently, and the overall interictal state of the epileptogenic region in this disorder is one of relatively decreased activity, as reflected in the hypometabolism seen on PET using 18F-fluorodeoxyglucose.61 It would appear therefore that the synaptic reorganization observed in human hippocampal sclerosis are associated with a chronic state of relative quiescence, but a propensity for hypersynchronous excitatory and inhibitory discharges.
It has been suggested that hippocampal reorganization alone, and the resultant repetitive hypersynchronous ictal discharges limited to the hippocampus, is not sufficient for the clinical manifestation of epileptic seizures. In fact, these electrographic ictal phenomena can be recorded with depth
electrodes, unassociated with clinical signs or symptoms.108 Complex partial, and perhaps even simple partial, seizures would seem to require propagation of ictal discharge to adjacent and distant structures, which might be facilitated if these structures were particularly receptive and themselves epileptogenic. Secondary epileptogenesis, as occurs with kindling and the mirror focus phenomenon,88 could conceivably account for transsynaptic changes necessary for behaviorally apparent ictal events to ensue. In primates, bilateral hippocampal lesions may be necessary before temporal lobe seizures can be observed,107 and in patients with medically refractory TLE, some degree of bilateral mesial temporal epileptogenicity is the rule rather than the exception.119 Patients with mesial TLE and hippocampal sclerosis often experience the recurrence of auras after radical anterior temporal removal has succeeded in abolishing the habitual complex partial seizures. Perhaps the epileptiform region is widespread from the beginning, or perhaps secondary epileptogenicity results in extension to the ipsilateral posterior hippocampus, contralateral hippocampus, or wide areas of limbic cortex in either hemisphere.
electrodes, unassociated with clinical signs or symptoms.108 Complex partial, and perhaps even simple partial, seizures would seem to require propagation of ictal discharge to adjacent and distant structures, which might be facilitated if these structures were particularly receptive and themselves epileptogenic. Secondary epileptogenesis, as occurs with kindling and the mirror focus phenomenon,88 could conceivably account for transsynaptic changes necessary for behaviorally apparent ictal events to ensue. In primates, bilateral hippocampal lesions may be necessary before temporal lobe seizures can be observed,107 and in patients with medically refractory TLE, some degree of bilateral mesial temporal epileptogenicity is the rule rather than the exception.119 Patients with mesial TLE and hippocampal sclerosis often experience the recurrence of auras after radical anterior temporal removal has succeeded in abolishing the habitual complex partial seizures. Perhaps the epileptiform region is widespread from the beginning, or perhaps secondary epileptogenicity results in extension to the ipsilateral posterior hippocampus, contralateral hippocampus, or wide areas of limbic cortex in either hemisphere.
Related posts:
 Epidemiology of Acute Symptomatic Seizures
EEG Traits
Voltage-Gated Ion Channels: Molecular Biology and Role of Mutations in Epilepsy
Basal Ganglia and Brainstem Anatomy and Physiology
The Effect of Seizures on the Brain
Typical and Atypical Absence Seizures, Myoclonic Absences, and Eyelid Myoclonia
Epidemiology of Acute Symptomatic Seizures
EEG Traits
Voltage-Gated Ion Channels: Molecular Biology and Role of Mutations in Epilepsy
Basal Ganglia and Brainstem Anatomy and Physiology
The Effect of Seizures on the Brain
Typical and Atypical Absence Seizures, Myoclonic Absences, and Eyelid Myoclonia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


