Basic Principles of Medicinal Chemistry
Donald F. Weaver
Raman Sankar
Introduction
Medicinal chemistry is the branch of applied organic chemistry that deals with the design and synthesis of new chemical entities for the symptomatic or curative treatment of human disease states.30 Medicinal chemistry is a multidisciplinary endeavor. It has as its basis a firm understanding of all branches of chemistry (in particular synthetic organic chemistry), but also embraces many elements of other disciplines that are necessary tools for the practicing medicinal chemist, including pharmacology, pharmacy, physiology, biochemistry, and medicine. The primary objective of medicinal chemistry is to discover new prototype molecules as prospective drugs, and subsequently to optimize any promising drug candidate by improving its beneficial therapeutic effects and minimizing its undesirable side effects. As applied to epilepsy, the role of medicinal chemistry is to discover new drugs to treat seizures with improved efficacy and decreased toxicity.
The medicinal chemistry of anticonvulsants has a long history, and it will probably have a long future. During the past 2,000 years, many compounds have been suggested as potential therapeutic agents for epilepsy. The history of the medicinal chemistry of anticonvulsant drugs can be categorized into three distinct eras: The era of charlatanism (prehistory to 1857), the era of serendipity (1857 to 1980), and the era of rational drug design (1980 to the present). The future of anticonvulsant medicinal chemistry will also hold a number of distinct changes.
The Era of Charlatanism: Ancient Times to 1857
Epilepsy was described by the school of Hippocrates in the book On the Sacred Disease, written in 400 BC. Although the role of heredity as well as the relationship between focal head trauma and contralateral seizures was recognized at the time, the overwhelming emphasis on the role of possession by evil spirits and excess accumulation of cold phlegm resulted in therapeutic approaches that focused on occult or magical powers to render a cure. The therapy of that time was dominated by superstition and charlatanism and relied on agents such as gladiator blood, seal penis, mistletoe, and dried human skull extract. Not surprisingly, such cures were of negligible value.
It was not until the “era of serendipity” that truly useful chemical agents to treat seizures first emerged.
The Era of Serendipity: 1857 to 1980
Many important drugs have had their origin in an accident, either in the laboratory or the clinic. The so-called serendipity factor looms large in the history of medicinal chemistry and accounts for several major discoveries, from penicillin as an antibacterial agent to imipramine as an antidepressant. Although this is a humbling realization, the evolution from penicillin to the large number of synthetic β-lactams and cephalosporins in use today is a direct result of the application of concepts of drug design. Design concepts that build on a serendipitous lead to generate a large number of synthetic analogs have played a role in the development of several anticonvulsant drugs. One of the authors30 has stressed the critical importance of serendipity in a historical analysis of antiepileptic drug development, urging investigators to look sideways for the unexpected. From 1857 to 1980, serendipity was the major discoverer of anticonvulsant drugs.
The first application of effective chemotherapy to control seizures was undertaken by Sir Charles Locock, an obstetrician in the mid-19th century. In those days, bromide preparations were known for their sedative and antiaphrodisiac properties, and epilepsy was thought to result from excessive masturbation and hysteria. These factors caused Locock to introduce bromides for the treatment of seizures related to menses and seizures considered to be venereally induced. Although his reasoning was quite erroneous, this inorganic agent was the first effective pharmacotherapeutic agent for epilepsy.
The first synthetic organic medicinal agent to control seizures was phenobarbital, a ureide (a derivative of urea, synthesized by the combination of urea and substituted malonic acid) that was introduced in 1912.15 The discovery of the anticonvulsant potential of phenobarbital was quite accidental, as the drug had been administered mainly to sedate a ward of noisy epileptic patients during the night. From that point on, the increasing ability of chemists to tinker with the basic ring structure of phenobarbital, combined with new animal models of seizures, became the driving force behind the development of anticonvulsant drugs.
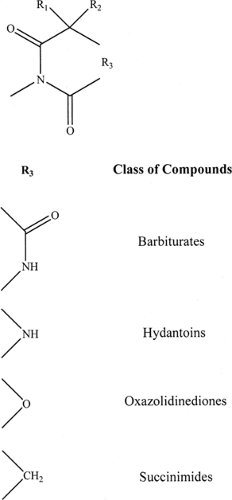 FIGURE 1. Structure common to antiepileptic drugs containing the ureide moiety. |
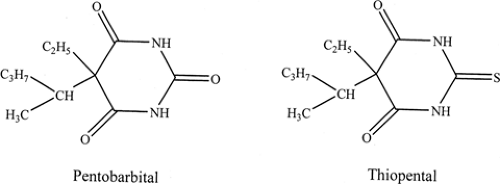 FIGURE 2. Substitution of the urea oxygen on pentobarbital with sulfur to create thiopental results in a compound with increased lipid solubility; this in turn leads to extremely rapid distribution of thiopental into the central nervous system and rapid redistribution to adipose tissue, thus making it a member of the ultra-short series. |
The first epochal result of such an approach was the discovery of phenytoin by Merritt and Putnam.19 The development of the maximal electroshock model by Merritt and Putnam,20 with its subsequent use as a screening model, was crucial to the availability of phenytoin in 1938. The close structural relationship between phenobarbital (a barbiturate) and phenytoin (a hydantoin) can be discerned from FIGURE 1. The expansion of the barbiturate and hydantoin families came about because the side chains (aryl, alkyl, cycloalkyl) could be varied and N-alkyl groups could be introduced to the basic structure of phenobarbital and phenytoin, giving rise to a number of compounds that differed in their onset and duration of action. The subsequent availability of the pentylenetetrazol model gave rise to the development of trimethadione, an oxazolidindione, as a specific agent for absence (then call petit mal) seizures. This compound was originally studied as an analgesic; it was systematic screening using available animal models that resulted in its accidental discovery as an antiabsence drug. The development of the succinimides, especially ethosuximide, yielded effective antiabsence compounds that were also less toxic. The close structural
relationship among the barbiturates, hydantoins, oxazolidindiones, and succinimides (families of compounds in the ureide dynasty), all of which possess a cyclic structure, is clearly seen in FIGURE 1. All these compounds resulted from the serendipitous clinical observation of phenobarbital as an anticonvulsant.
relationship among the barbiturates, hydantoins, oxazolidindiones, and succinimides (families of compounds in the ureide dynasty), all of which possess a cyclic structure, is clearly seen in FIGURE 1. All these compounds resulted from the serendipitous clinical observation of phenobarbital as an anticonvulsant.
A wealth of lessons in medicinal chemistry was learned from the initial studies with ureides. For example, it was noted that the lipid solubility of a given compound within the ureide family could be increased according to the size of the alkyl (cycloalkyl) and aryl side chains, by the introduction of N-alkyl groups (which alter the pKa of the compound and hence the extent of ionization), or by replacing an oxygen in the ureide ring with a sulfur (Fig. 2).
The lipid solubility of a compound correlates with the rapidity with which therapeutic levels may be achieved in the brain. Other chemical modifications influenced the rate of drug clearance by affecting plasma protein binding, affinity for the hepatic cytochrome P450/mixed-function oxidase system, and the rate of the oxidative reaction in that matrix. Accordingly, the structural congeners developed via medicinal chemistry gave rise to drugs with distinctive onsets of action as well as duration of action.
The ureides dominated the anticonvulsant drug scene until the arrival of the benzodiazepines, carbamazepine, and valproic acid. All three of these classes of drug were discovered by serendipity. For example, chlordiazepoxide was discovered when it was accidentally synthesized in an attempt to produce antipsychotic tricyclic drugs; its sedative and anxiolytic properties were then observed serendipitously, and only later were the anticonvulsant properties of benzodiazepines realized. Similarly, carbamazepine was designed as a psychotropic tricyclic; its unanticipated anticonvulsant properties were revealed at a later time.
However, the importance of serendipity in drug discovery is most clearly illustrated by the circumstances in which the anticonvulsant activity of valproate was recognized. In 1963, a graduate student in France prepared a series of heterocyclic molecules that were subsequently submitted to a local pharmaceutical company for screening in a variety of biologic assays. Because of the poor solubility of these compounds, they were somewhat randomly dissolved in a series of solvents, of which valproic acid was one. When it was noted that one of the compounds seemed to have anticonvulsant activity, subsequent work revealed that all such activity was located in the serendipitously selected valproic acid solvent.4,5 Thus, valproate was discovered completely by accident.
As is apparent, the major traditional anticonvulsant drugs (phenobarbital, phenytoin, carbamazepine, valproate, benzodiazepines) were all discovered through serendipitous observations. Because of this element of chance, the medicinal chemistry of these important compounds remains incomplete. For example, modern electrophysiologic studies suggest that phenytoin, carbamazepine, and valproate probably function at the level of the voltage-gated sodium channel; however, the structure of the receptor site, the nature of the drug–receptor interaction, and the portion of the drug molecule interacting with the receptor all remain incompletely elucidated.
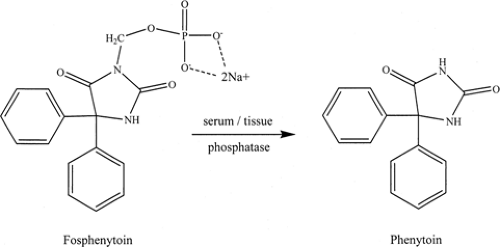 FIGURE 3. Modern antiepileptic drugs. |
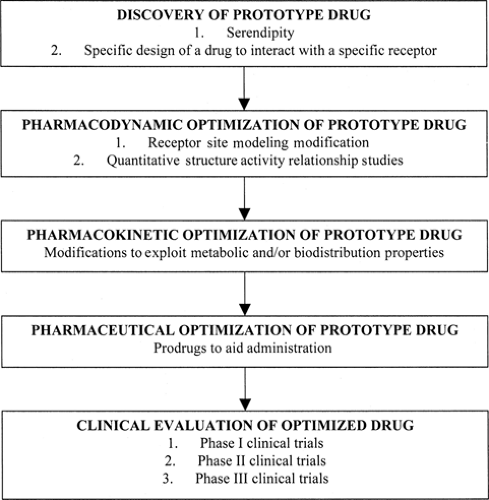 FIGURE 4. Modern antiepileptic drug design and optimization. |
The Era of Rational Drug Design: 1980 to the Present
Over the past 25 years, a number of new chemical entities have been introduced as new agents for the treatment of epilepsy, with many more on the drawing board.18 Some of these new agents are shown in FIGURE 3. This past quarter-century has been an era of rational drug design.
Strictly defined, rational drug design is a process whereby the three-dimensional structure of some biologic macromolecular receptor involved in the etiology and pathogenesis of a given disease process is determined, thus permitting the intelligent engineering of molecules specifically developed to dock with and alter the function of the receptor macromolecule. Operationally defined, rational drug design is the process of optimizing a prototype drug to yield the best possible clinical candidate. A flow chart describing this process is shown in FIGURE 4.
Rational drug design starts with the identification of a prototype or lead compound. This may be done using several approaches, including random screening and specific design. The
lead compound is then optimized at three distinct levels: Pharmacodynamic, pharmacokinetic, and pharmaceutical. At the pharmaceutical level, the route of administration and the adequacy of solubility and penetration into the organism are determined. The pharmacokinetic level concerns the metabolism of the drug (including first-pass effect through the liver) and its biodistribution to the receptor microenvironment. The pharmacodynamic phase involves the interaction of the drug with its receptor at the molecular level of conceptualization.
lead compound is then optimized at three distinct levels: Pharmacodynamic, pharmacokinetic, and pharmaceutical. At the pharmaceutical level, the route of administration and the adequacy of solubility and penetration into the organism are determined. The pharmacokinetic level concerns the metabolism of the drug (including first-pass effect through the liver) and its biodistribution to the receptor microenvironment. The pharmacodynamic phase involves the interaction of the drug with its receptor at the molecular level of conceptualization.
Discovering the Prototype Drug
Craig6 described several methods of “drug discovery” to convey the fact it is usually not restricted to de novo drug design. The general approaches to the identification of prototype compounds include the three described below:
Specific design of a drug to interact with a receptor site after the structure of the receptor site has been rigorously determined by x-ray crystallography or other means
Preparation of structural analogs of synthetic chemicals with interesting biologic activity
Preparation of novel chemicals based on the known structures of naturally occurring, biologically active substances of plant and animal origin
Approach 1: This ultimate ideal in rational drug design involves the isolation, purification, and crystallization of receptor proteins. As many of the relevant antiseizure receptor targets (such as the transmembrane voltage-gated sodium channel) are not amenable to such biochemical manipulations, this approach remains unrealized as far as anticonvulsant drug discovery is concerned. Nevertheless, ion channels remain as an important target for anticonvulsant drug design.31 Also, recent pioneering work by McKinnon (for which he was awarded the 2003 Nobel Prize in Chemistry) is starting to provide insights into the structure of voltage-gated ion channels.
Approach 2: For years it was believed that folate deficiency was a mechanism of anticonvulsant activity. For instance, many workers suggested that the mechanism of phenytoin might be exerted through this route. Using this as a starting point, attempts were made to design novel anticonvulsant drugs as analogs of compounds with known antifolate activity. Lamotrigine, which was developed as a congener of methotrexate, demonstrated significant anticonvulsant activity. Unfortunately, the success of lamotrigine as an anticonvulsant drug does not occur via a folate mechanism, but rather through a sodium channel blockade—the importance of the serendipity factor persists even in the era of rational drug design.
Approach 3: In rational drug design, discovering a prototype drug usually requires that the underlying biochemistry be understood at some reasonable level of refinement. Seizures occur when an imbalance in the two principal neurotransmitters occurs: L-glutamic acid, an excitatory amino acid neurotransmitter, and γ-aminobutyric acid (GABA), an inhibitory amino acid neurotransmitter. Of these two neurotransmitters, GABA is particularly important in epilepsy. The concentration of GABA is regulated by two pyridoxal-5-phosphate (PLP)-dependent enzymes: L-glutamic acid decarboxylase (GAD, glutamate decarboxylase), which converts glutamate to GABA, and GABA aminotransferase (GABA-T), which catabolizes GABA to succinic semialdehyde. (Although succinic semialdehyde is cytotoxic, it is rapidly and efficiently metabolized to succinic acid by the enzyme succinic semialdehyde dehydrogenase [SSADH].)
Because GABAergic dysfunction seems to play a role in the pathogenesis and etiology of seizures and epilepsy, it seems understandable that GABA or some related analog might be a profitable prototype drug. A wealth of experimental data supports this assertion. For example, when GABA levels fall below a certain threshold level, seizures may occur; likewise, direct injection of GABA into the brains of experimental animals can stop seizure activity. Accordingly, it would seem reasonable that GABA should be an ideal prototype anticonvulsant drug. Nevertheless, when GABA is administered orally, it is devoid of anticonvulsant activity, as it is not bioavailable to the brain; GABA does not cross the blood–brain barrier. Another approach for increasing brain GABA levels would be to discover a compound capable of crossing the blood–brain barrier and inactivating the GABA-T enzyme implicated in the biodegeneration of GABA. Provided the glutamate decarboxylase was not simultaneously inhibited, GABA levels would rise to effective anticonvulsant concentrations. Vigabatrin, a structural analog of GABA, is such a molecule. Vigabatrin is a molecule that functions as a mechanism-based inactivator of GABA-T. Vigabatrin is an unreactive compound that is converted by the normal catalytic mechanism of GABA-T to a reactive intermediate compound that attaches to the GABA-T, thereby inactivating the enzyme.
It is surprising that GABA does not cross the blood–brain barrier, but vigabatrin, which is also a small, charged molecule, is capable of diffusing across the lipophilic blood–brain barrier. There are two reasons for this seeming paradox. First, the attachment of a vinyl substituent increases the lipophilicity of the molecule. Second, the vinyl group is an electron-withdrawing substituent that lowers the pKa of the amino group; this in turn increases the concentration of the uncharged, nonzwitterionic form, which is more lipophilic than the charged, zwitterionic form.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


