Benzodiazepines
Dieter Schmidt
Alan J. Wilensky
Introduction
The benzodiazepines represent a clinically relevant class of compounds that possess a broad range of properties, including anticonvulsant efficacy. In 1965, diazepam was the first benzodiazepine used to treat status epilepticus (SE) in humans and, since then, several other benzodiazepines have entered the market.29 Currently, benzodiazepines such as diazepam or lorazepam are drugs of first choice for emergency treatment of acute seizures, series of seizures, and initial treatment of SE.56 More recently, buccal midazolam has been evaluated for emergency use in acute seizures, mainly in children.44 In addition, a number of other benzodiazepines such as clobazam, clonazepam, clorazepate, and nitrazepam are in clinical use in various parts of the world, mostly as add-on medication for refractory epilepsy. However, sedation and hypersalivation at doses that are comparable to those that protect against seizures, and, importantly, loss of efficacy during prolonged treatment, have limited their usefulness. Earlier reviews are available.29,57 This chapter outlines the chemistry, pharmacology (including clinical pharmacokinetics), efficacy, adverse effects, and clinical use of several benzodiazepines—clorazepate, clobazam, clonazepam, diazepam, lorazepam, midazolam, and nitrazepam—used in the treatment of epilepsy.
Chemical Structure, Formulations and Methods for Determination in Body Fluids
The term benzodiazepine refers to that central portion of the chemical structure comprising a benzene ring attached to a seven-membered diazepine ring (Fig. 1A). The majority of clinically important benzodiazepines contain a third ring (5-aryl substituent) and have nitrogen substitutions at the 1 and 4 positions of the diazepine ring (Fig. 1B); these are generally referred to as 1,4-benzodiazepines. Altering the positions of the two nitrogen atoms in the heterocyclic ring can confer similar anticonvulsant properties. An example is clobazam, a 1,5-benzodiazepine.2,29 Both midazolam, a short-acting benzodiazepine, and flumazenil, an equally short-lived benzodiazepine competitive antagonist, have imidazole rings attached at position 1 of the diazepine ring, in contrast to other anticonvulsant benzodiazepines, which have hydrogen or an alkyl group in that position.2
Formulations
In many countries—with the notable exception of the United States—clobazam is available for oral administration as tablets but not available for intravenous or intramuscular injection. Clonazepam can be administered as tablets and drops and as an intravenous formulation. Clorazepate can be prescribed as tablets and slow-release capsules and, in some countries, as an intravenous and intramuscular formulation. Diazepam is administered by oral, intravenous, intramuscular, and rectal routes and is available as oral solution, tablets, a sustained-release capsule, a rectal suppository, a rectal solution, a rectal gel, and a parenteral formulation. Intramuscular injection of diazepam produces variable serum levels and is not a recommended route of administration.57 Lorazepam is widely available as oral and sublingual tablets and as an intravenous and intramuscular formulation. Midazolam is in use as a tablet, syrup, and a parenteral formulation for buccal, oral, intranasal, and rectal routes. Nitrazepam is administered by oral, rectal, and intravenous and intramuscular routes.2
Methods for Determination of Benzodiazepines in Body Fluids
Assay methods are available for the determination of serum levels of various benzodiazepines, including gas and liquid chromatographic procedures.2,29 However, as will be pointed out, their usefulness for guiding the treatment of epilepsy is limited, mainly because the dosage is usually titrated against immediate therapeutic effects, and the correlation of serum concentration to effect during chronic treatment is hampered by the development of tolerance to therapeutic and adverse effects of benzodiazepines.
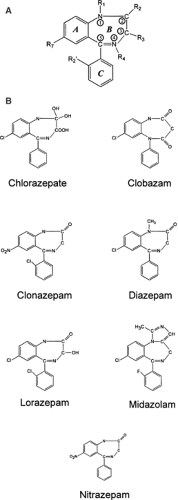 FIGURE 1. A: The generic chemical structure of benzodiazepines. B: The chemical structure of common anticonvulsant benzodiazepines. Note that clobazam is a 1,5-benzodiazepine. |
Pharmacology
Activity in Experimental Models of Seizures/Epilepsy
Benzodiazepines effectively block seizures induced by pentylenetetrazol, a potent noncompetitive antagonist at γ-aminobutyric acid (GABA)A receptors. Activity in this animal seizure model has been considered very predictive of drugs with potential clinical utility against myoclonic and absence seizures.39 Benzodiazepines also block seizures induced by maximal electroshock model, which is used to screen possible antiepileptic drugs (AEDs) for the treatment of partial seizures with or without secondary generalization. In addition, the benzodiazepines have been shown to be effective in models of partial seizures induced by cortical lesions induced by a variety of agents, including alumina cream, cobalt, penicillin, and strychnine.39 Finally, loss of efficacy during prolonged treatment with benzodiazepines has been shown in a number of experimental models of partial epilepsy, including amygdala kindling.37
Mechanisms of Action
The primary target of benzodiazepines is a postsynaptic receptor for the neutral amino acid GABA, the major inhibitory neurotransmitter in the central nervous system (CNS). GABA is released from presynaptic neurons and binds to at least two classes of receptors, GABAA and GABAB receptors, which are found on almost all cortical neurons.40 It is well established that benzodiazepines have primary actions as anticonvulsants by interacting with GABAA receptors at the benzodiazepine binding site and allosterically modifying the GABA receptor currents to enhance inhibition.40 Binding to the benzodiazepine recognition site results in enhancement of GABAA receptor current by increasing the frequency of the GABAA receptor opening without significantly affecting the mean open time or conductance of the channel.40 This is in contrast to barbiturates, which increase the mean open time and burst duration of GABAA receptor channel openings without affecting frequency of channel opening.40,51 It is important to recognize that benzodiazepines, unlike barbiturates, cannot directly activate the GABAA receptor in the absence of GABA, the endogenous ligand, even at very high concentrations.40 However, advances in understanding the molecular biology of GABA receptors demonstrate that the GABA receptor is made up of many different subunits (α, β, γ, δ, ρ) with different subtypes in many combinations; it is undergoing continual characterization.40 GABAA receptors containing the α1 subunit are an important target for protection of seizures by benzodiazepines.51 The effects of benzodiazepines on tonic GABAA receptor currents, which originate from GABA acting on extrasynaptic receptors, may be important for their action.51 The antiabsence activity of benzodiazepines probably results from their ability to interfere with hypersynchronous activity in the thalamocortical circuitry that is believed to underlie the 3-Hz spike-and-wave activity that is characteristic of generalized absence seizures.51 More specifically, benzodiazepines are thought to reduce the inhibitory output of the reticular neurons by effects on benzodiazepine-sensitive α3-containing GABAA receptors and therefore prevent absence seizure activity.51
It is of clinical interest that the multiple GABA receptor isoforms described here have a differential sensitivity to benzodiazepine receptor ligands that may have different clinical actions. In the future, it may be possible to develop benzodiazepine receptor ligands that have selective anticonvulsant actions without undesirable side effects. One such side effect is the development of tolerance to effect during prolonged treatment. Although the exact mechanism of tolerance is not clear, it has been suggested to include a reduction in the allosteric interactions between the GABAA receptors at the benzodiazepine recognition site and changes in the subunit pattern.49 Because tolerance to adverse effects such as sedation sets in within days, whereas tolerance to efficacy takes weeks to months to fully develop, the mechanisms for tolerance to side effects and efficacy may differ. However, some of the clinical effects of benzodiazepines in experimental animals cannot be explained simply by their potentiation of GABA-activated currents,29 and benzodiazepines may act on targets other than the GABAA receptor. Through unknown mechanisms, benzodiazepines may produce elevations of GABA levels in the cerebrospinal fluid.38 At high concentrations, especially those that may be achieved in the treatment of SE, benzodiazepines may have significant effects on inhibiting currents carried by voltage-gated sodium and, to a lesser extent, calcium channels.40
Table 1 Clinical pharmacokinetics of benzodiazepines | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
Clinical Pharmacokinetics
Absorption
In general, the benzodiazepines are absorbed rapidly, with peak concentrations after oral use ranging from 0.5 to 4 hours. The absorption of orally administered benzodiazepines is 80% or more.2
Although clorazepate is soluble and well absorbed orally, the acidic gastric environment is bypassed during intravenous administration, resulting in less production of the active metabolite desmethyldiazepam. Thus, intravenous administration is not useful in the treatment of SE.2
Within tens of seconds after intravenous administration, the anticonvulsant effect of diazepam can be seen on an
electroencephalogram (EEG) as suppression of epileptiform discharges in humans.57 On a cautionary note, intravenous diazepam solution may precipitate when mixed with saline solution, and it may also adsorb to polyvinyl tubing.29,56 If left diluted in an intravenous fluid bag for 24 hours, diazepam is reported to lose 95% of its intrinsic activity;29 thus, fresh solutions should be made within 6 hours of use. Rectal delivery of diazepam results in higher and earlier peak blood concentrations compared with either oral or intramuscular routes.
electroencephalogram (EEG) as suppression of epileptiform discharges in humans.57 On a cautionary note, intravenous diazepam solution may precipitate when mixed with saline solution, and it may also adsorb to polyvinyl tubing.29,56 If left diluted in an intravenous fluid bag for 24 hours, diazepam is reported to lose 95% of its intrinsic activity;29 thus, fresh solutions should be made within 6 hours of use. Rectal delivery of diazepam results in higher and earlier peak blood concentrations compared with either oral or intramuscular routes.
Lorazepam can be administered orally, sublingually, and intravenously. It is rapidly distributed into the brain, and peak levels are attained within 5 minutes.2 Human studies demonstrate sedative effects and EEG sleep spindles within 0.5 to 4 minutes of infusion of doses ranging between 2 and 5 mg.29 The ability to administer lorazepam sublingually is attractive when intravenous access is not readily available.24 Sublingual use of lorazepam is easier to administer, maintains privacy, and is seen as more dignified than rectal application of diazepam. Some physicians recommend it instead of rectal diazepam for prevention of seizure recurrence. Sublingual lorazepam was successful in controlling serial seizures in ten children.69
Plasma Protein Binding and Distribution
The various benzodiazepines are fairly highly protein-bound, although it is the free (unbound) benzodiazepine that is active (Table 1). The benzodiazepines as a group are lipophilic and have a high volume of distribution. In obese patients, the volume of distribution is larger.29 Some studies show a larger volume of distribution of benzodiazepines in female patients compared with that in male patients, which is thought to be a reflection of the proportion of adipose tissue.29 Brain levels of diazepam are nearly double that of serum concentrations, and appear to be highest in brainstem and white matter compared with gray matter.29 Protein binding is reduced in neonates and patients with hepatic disease. The mean apparent volume of distribution is approximately 1.1 L/kg (range, 0.8 to 2.6 L/kg), and is generally higher in young children.29 Renal or hepatic disease results in a decreased plasma clearance of desmethyldiazepam. Diazepam distributes rapidly throughout lipid tissues and quickly crosses the blood–brain barrier (BBB).29 Within minutes, however, brain concentrations decline swiftly as diazepam is redistributed to other fatty tissues, resulting in a short distribution half-life (tα, approximately 1 hour).29,57 After a single bolus injection, the concentration of free (active) diazepam in the brain may fall below therapeutic levels within half an hour.57
Metabolism
All the anticonvulsant benzodiazepines, except for lorazepam and clonazepam, are metabolized by hepatic microsomal enzymes to pharmacologically active demethylated and hydroxylated compounds26,29 (Fig. 2). These metabolites are subsequently glucuronidated to inactive, more water-soluble compounds that are readily excreted in the urine. The active metabolites are often very important; for example, clorazepate is essentially a prodrug for its active metabolite, desmethyldiazepam. Clobazam has up to 14 metabolites, but the main active metabolite is desmethylclobazam, also called norclobazam. Desmethylclobazam has comparable affinity for the brain benzodiazepine receptor and also has antiepileptic activity.57 After long-term administration, at steady state the serum concentration of desmethylclobazam in most studies is eight to 20 times higher than that of clobazam.29 Clobazam and its metabolite desmethylclobazam both have anticonvulsant properties, but desmethylclobazam is only one-fifth as potent as the parent compound.29 Because the ratio of plasma levels of the metabolite desmethylclobazam to those of the parent drug is about 10:1, and because the metabolite also has a longer half-life, the anticonvulsant property may be more attributable to desmethylclobazam than to clobazam.
Clonazepam undergoes nitroreduction to 7-aminoclona-zepam, followed by acetylation to 7-acetamidoclonazepam, but neither metabolite appears to have clinically important pharmacologic activity.25 A major metabolic pathway for clonazepam involves acetylation. Rapid acetylators are more likely than slow acetylators to require higher doses.
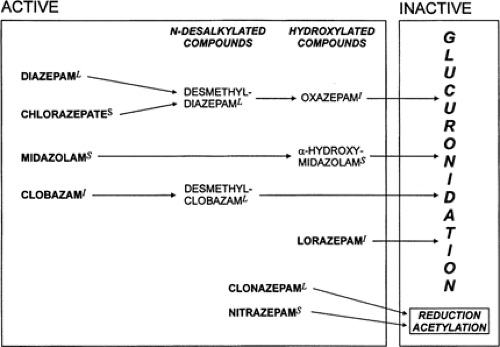 FIGURE 2. The metabolism and biotransformation of anticonvulsant benzodiazepines. |
Clorazepate is a prodrug and exerts its effects through its active metabolite, desmethyldiazepam. Clorazepate undergoes complete transformation (100%) to desmethyldiazepam.29 Desmethyldiazepam is also the main metabolite of diazepam
(see section on diazepam for a description of its characteristics). Clorazepate has intrinsic anticonvulsant activity, but this is small compared with that of desmethyldiazepam. Clorazepate is decarboxylated to desmethyldiazepam mainly in the stomach and bloodstream. The decarboxylation rate of clorazepate to desmethyldiazepam in the stomach is pH-dependent, with a lower pH resulting in a faster rate of conversion. There is a relatively instantaneous breakdown of clorazepate to desmethyldiazepam at a pH of 4 (90% at 10 minutes and 97% at 1 hour).29 Desmethyldiazepam rapidly crosses the BBB. Desmethyldiazepam is hydroxylated to oxazepam, which is conjugated fairly rapidly and excreted; thus, desmethyldiazepam has no active metabolites.
(see section on diazepam for a description of its characteristics). Clorazepate has intrinsic anticonvulsant activity, but this is small compared with that of desmethyldiazepam. Clorazepate is decarboxylated to desmethyldiazepam mainly in the stomach and bloodstream. The decarboxylation rate of clorazepate to desmethyldiazepam in the stomach is pH-dependent, with a lower pH resulting in a faster rate of conversion. There is a relatively instantaneous breakdown of clorazepate to desmethyldiazepam at a pH of 4 (90% at 10 minutes and 97% at 1 hour).29 Desmethyldiazepam rapidly crosses the BBB. Desmethyldiazepam is hydroxylated to oxazepam, which is conjugated fairly rapidly and excreted; thus, desmethyldiazepam has no active metabolites.
Diazepam is biotransformed by hepatic microsomal enzymes in two principal steps: initial demethylation to desmethyldiazepam, followed by hydroxylation to oxazepam (see Fig. 2). Oxazepam is then conjugated to glucuronide, producing an inactive metabolite that is largely excreted in the urine.
Desmethyldiazepam, which is also an active metabolite of clorazepate (see section on clorazepate), has one-third the anticonvulsant potency of its parent compound, diazepam.21 Biotransformation is also age-dependent, with limited demethylation and hydroxylation beginning as early as the 13th week of gestation, and enzymatic capacity increases steadily to maturity during the first few years of life.
Lorazepam is rapidly converted by hepatic metabolism to a glucuronide through conjugation at the 3-hydroxyl portion of the seven-membered diazepine ring.28 More than 75% of the drug is converted to the glucuronide and excreted in the urine. The glucuronide is an inactive metabolite and does not have any toxic or antiepileptic effects. Two minor metabolites have no significant toxic or antiepileptic activity.28 Hepatic failure results in the accumulation and reduced clearance of the parent drug, lorazepam, resulting in prolongation of the elimination half-life of lorazepam by 50%.28 Renal failure does not affect clearance of the parent drug; however, it causes accumulation and reduced clearance of the glucuronide metabolite, but no additional toxicity.28,29
Midazolam metabolism occurs by oxidation of the imidazole ring, mostly to 1-hydroxymidazolam, which has about 10% of the biologic activity of midazolam and an elimination half-life of 1 hour.2
The major metabolic pathway of nitrazepam involves hepatic nitroreduction, with 7-aminonitrazepam and 7-acetami-nonitrazepam being the principal metabolites. The metabolites are inactive and are excreted renally. Only about 1% of the unchanged nitrazepam shows up in the urine.29 Glucuronidation is less affected by aging or hepatic disease than demethylation or hydroxylation. Thus, lorazepam and midazolam would be preferable among benzodiazepines for elderly patients or individuals with cirrhosis. Nevertheless, caution must always be exercised in prescribing drugs to older patients, who have a generally diminished volume of distribution and decreased metabolism.22
Elimination
Clonazepam and clobazam and its metabolite desmethylclobazam are slowly eliminated, with a half-life of 20 to 56 hours (see Table 1). Because of its long half-life, alternate-day administration of clonazepam or clobazam is possible and has been suggested to reduce tolerance without sacrificing seizure control. Because of the long half-life of desmethyldiazepam, clorazepate can be given once a day, but divided doses decrease the likelihood of toxicity and allow for more stable steady-state levels than once-a-day dosing.66 Clearance of unbound desmethyldiazepam was reduced in renal failure, although the free fraction was increased and the elimination half-life (tβ) was shortened compared with those of healthy controls, highlighting the complex nature of drug pharmacokinetics in certain disease states.29 Severe liver disease seems to prolong the half-life of desmethyldiazepam.
The tβ of diazepam ranges from 1 to 2 days in healthy volunteers and is age-dependent—shorter in infants and children, and prolonged in premature infants and the elderly.57 Desmethyldiazepam has a much longer tβ than diazepam, because its formation depends on the continued redistribution of diazepam from peripheral to central compartments, and back again to the periphery. Thus, the tβ for desmethyldiazepam is longer with administration of diazepam than of clorazepate, a prodrug completely converted to desmethyldiazepam. This results in a greater accumulation of desmethyldiazepam than diazepam in both plasma and cerebrospinal fluid during long-term treatment.29
For clinical use, the duration of effect of various benzodiazepines is an important variable. The “long-acting” agents, such as clobazam, clorazepate, and lorazepam, have half-lives longer than 24 hours and produce active metabolites;
the “intermediate-acting” benzodiazepines, such as nitrazepam and clonazepam, have half-lives shorter than 24 hours and do not produce active metabolites; there are also “short-acting” agents, such as midazolam.26 Two factors must be taken into consideration regarding duration of effect: distribution half-life (tα) and tβ. During acute administration, as in the treatment of SE, the rates of absorption and distribution are important, whereas during long-term therapy, in which distribution is complete, the tβ is more critical, because it largely determines steady-state serum levels of a drug. This principle is best illustrated by the clinical use of diazepam and lorazepam. Oral diazepam has a short tα (approximately 1 hour) but a long tβ (approximately 40 hours); furthermore, the major active metabolite, desmethyldiazepam, prolongs the tβ to 100 hours or more. The net result after a single dose, because of rapid distribution, is a short duration of action. After repeated doses, diazepam and its metabolites potentially accumulate in the body. Oral lorazepam, on the other hand, has a longer tα (1.5 hours) than diazepam but a shorter tβ (14 hours), and it produces no active metabolites.23 Thus, compared with diazepam, a single dose of lorazepam leads to a longer lasting clinical effect, and repeated doses of lorazepam are less likely to produce very high levels throughout the body.
the “intermediate-acting” benzodiazepines, such as nitrazepam and clonazepam, have half-lives shorter than 24 hours and do not produce active metabolites; there are also “short-acting” agents, such as midazolam.26 Two factors must be taken into consideration regarding duration of effect: distribution half-life (tα) and tβ. During acute administration, as in the treatment of SE, the rates of absorption and distribution are important, whereas during long-term therapy, in which distribution is complete, the tβ is more critical, because it largely determines steady-state serum levels of a drug. This principle is best illustrated by the clinical use of diazepam and lorazepam. Oral diazepam has a short tα (approximately 1 hour) but a long tβ (approximately 40 hours); furthermore, the major active metabolite, desmethyldiazepam, prolongs the tβ to 100 hours or more. The net result after a single dose, because of rapid distribution, is a short duration of action. After repeated doses, diazepam and its metabolites potentially accumulate in the body. Oral lorazepam, on the other hand, has a longer tα (1.5 hours) than diazepam but a shorter tβ (14 hours), and it produces no active metabolites.23 Thus, compared with diazepam, a single dose of lorazepam leads to a longer lasting clinical effect, and repeated doses of lorazepam are less likely to produce very high levels throughout the body.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


