The toxin is initially synthesized as a single-chain polypeptide and is subsequently cleaved to yield a heavy chain linked by a disulfide bond to a light chain. After endocytosis, the disulfide bond is cleaved separating the chains. The N terminus of the light chain is the active proteolytic site responsible for cleavage of SNARE (SNAP (soluble NSF attachment protein) receptor) proteins and the resultant permanent inhibition of acetylcholine (ACh) release. Recovery occurs only when the axon sprouts another nerve terminal to re-establish synaptic transmission.
Clinical Features of Botulism
Exposure to botulinum toxin occurs through the following mechanisms:
1. Ingestion of pre-formed toxin (food-borne botulism)
2. Local production of toxin by C. botulinum organisms at the site of a penetrating wound (wound botulism)
3. Local production of toxin by C. botulinum in the gastrointestinal tract (infant botulism and adult intestinal botulism)
4. Inhalation of pre-formed toxin (inhalational botulism)
5. Inadvertent systemic exposure to the toxin during injection for therapeutic or cosmetic reasons (iatrogenic botulism).
The classic clinical presentation for botulism is an acutely evolving symmetrical, descending motor paralysis with autonomic symptoms in an alert patient, with no sensory deficits.
Classic Botulism (Food-Borne Botulism)
Almost all human cases of food-borne botulism are caused by A, B, and E strains. The geographical distribution parallels the distribution of spores with type A predominating in western USA and type B predominating in north-eastern and central USA. Marine food products account for 91% of type E outbreaks. Type A infections cause more severe illness. The most common sources of infection are home-canned (preserved) vegetables, fruit, and fish. Prevention of food-borne botulism requires killing the bacterium with heating to at least 120°C for 5 min, preferably in a high pressure setting. In contrast to the spores, the toxins are heat labile and inactivated by heating to 85°C for a minimum of 5 min.
The incubation period typically ranges from 12 h to 72 h. The illness may sometimes be so mild that no medical attention is sought, and not all people who ingest the contaminated food become symptomatic. The initial symptoms are typically gastrointestinal with nausea, vomiting, diarrhea, and abdominal cramping. Presenting neurological signs and symptoms are referable to extraocular and bulbar weakness and include blurring of vision, ptosis, diplopia, ophthalmoplegia, dysarthria, and dysphagia. This is followed by a characteristic bilateral, symmetric, descending paralysis with weakness of the upper limbs followed by truncal and lower extremity weakness and autonomic dysfunction. Respiratory compromise may occur due to upper airway obstruction and/or to diaphragmatic weakness. There are typically no sensory symptoms.
 tips and tricks
tips and tricks
- Botulism differs from other flaccid paralyses in that it always manifests initially with prominent cranial paralysis, it invariably has a descending progression, and there is an absence of sensory symptoms or signs.
- Prominent oculobulbar and facial weakness are early manifestations with pupillary abnormalities in 50%.
- Patients are afebrile unless a concurrent infection is present.
- Nausea, vomiting, and diarrhea often precede or accompany neurological manifestations in food-borne botulism.
Wound Botulism
Wound botulism is caused by infection of a contaminated wound with C. botulinum (a natural contaminant of soil throughout the USA), with subsequent absorption of locally produced toxin into the circulation. First reported almost exclusively in patients with traumatic and surgical wounds, it is more recently associated with injection drug users, and in particular with subcutaneous injection of heroin in a method called “skin popping,” designed to slowly release the drug. Infection is believed to result from C. botulinum spores contaminating the heroin. After injection, the spores germinate in an anaerobic tissue environment and release toxin. The presence of skin abscesses may suggest the diagnosis, but the clinical diagnosis is often challenging. Maxillary sinusitis associated with intranasal cocaine use has rarely been the source of wound botulism. The clinical syndrome of wound botulism closely resembles classic botulism, save for the absence of gastrointestinal symptoms. The diagnosis is established by detection of the organism in the wound or toxin from the circulation.
Infant Botulism
Infant botulism is caused by ingestion of C. botulinum spores, which colonize the gastrointestinal tract and produce toxin that is absorbed into the circulation. The source of spores in most cases is unknown, although the more common sources of infection for infants appear to be honey and environmental exposure. The age of onset is between 3 weeks and 8 months, with most cases occurring before the age of 6 years. The disease characteristically begins with lethargy and poor feeding, usually accompanied by constipation. This is typically followed by weakness of bulbar and limb muscles, hypotonia, loss of head control, poor sucking ability, and decreased movements. Autonomic involvement is common with constipation, tachycardia, hypotension, neurogenic bladder, and dry mouth.
Adult Intestinal Botulism
The pathogenesis of adult intestinal botulism is similar to that of infant botulism. Only a few cases have been recognized, and most have occurred postoperatively, or in adults with underlying pathology of the gastrointestinal tract causing an alteration of the normal gut flora, including prior antimicrobial therapy, achlorhydria, Crohn’s disease, or surgery. Diagnosis is established by isolation of the organism or toxin from fecal samples.
Inhalational Botulism
This form of botulism is caused by inhalation of aerosolized, pre-formed toxin which is absorbed into the circulation through the lungs. The use of aerosolized toxin as an agent of bioterrorism has the potential to lead to high numbers of casualties.
Iatrogenic Botulism
This refers to generalized weakness or autonomic dysfunction related to the use of botulinum toxin as a therapeutic agent.
Differential Diagnosis
The diagnosis should be suspected in an alert patient presenting with an acute onset of descending, painless paralysis without sensory involvement. Clinical suspicion may be increased in the case of an outbreak where there is a history of a common source of food exposure. The differential diagnosis for botulism may be systematically broken down into central, peripheral nerve, neuromuscular junction, and muscular causes as shown in Table 17.1.
Table 17.1. Diagnostic considerations in a patient presenting with acute weakness with prominent ocular/bulbar involvement
| Etiology | Distinguishing features/Ancillary tests |
| Central nervous system Brain-stem strokes Demyelination Rhombencephalitis/Infections Thiamin deficiency | Altered sensorium, long tract signs, upgoing toes, abnormalities of hearing, facial sensory loss; nystagmus and ataxia if present would be incompatible with botulism and suggest CNS/alternate etiology. Test: MRI of the brain, lumbar puncture |
| Anterior horn hell Poliomyelitis West Nile virus | Fever, meningeal signs. Classically asymmetric flaccid paralysis. CSF pleocytosis. West Nile serology |
| Peripheral nerve a. Guillian–Barré syndrome (GBS)a b. Miller-Fisher syndromea c. Heavy metal poisoninga d. Lyme disease e. Tick paralysis f. Diphtheria g. Porphyria h. Critical illness neuropathy/myopathy i. Sarcoidosis j. Marine poisoning: saxitoxin tetrodotoxin ciguatera toxin | a. GBS is usually associated with sensory symptoms at onset. NCS/EMG shows demyelination. LP: albuminocytological dissociation b. Ataxia, anti-GQ1b antibodies in Miller-Fisher syndrome c. Urine, blood heavy metal screen d. Lyme serology, CSF Lyme PCR, CSF studies e. Usually children. Search skin for tick exposure f. Check for tonsillar exudates, culture g. Porphyria screen h. Neuropathic/myopathic findings in light of severe systemic illness, steroid use i. Radicular pattern, sensory involvement. Chest radiograph/imaging, serum ACE levels, MRI of the brain and spine as indicated. CSF studies j. Prominent sensory symptoms, pure motor presentation is very unusual |
| Neuromuscular junction a. Myasthenia gravisa b. Lambert–Eaton syndromea c. Aminoglycosides, neuromuscular blocking agents (drugs, venoms, Mg2+) d. Organophosphate poisoning | a, b. Subacute/chronic onset. Areflexia may be seen in LES but not in MG. NCS/EMG may show a decrement with repetitive nerve stimulation in MG and LEMS. Low baseline CMAPs with prominent facilitation seen with the latter. Serological studies: anti-AChR, anti-MuSK, anti-voltage-gated calcium channel antibodies maybe helpful. Organophosphate poisoning shows prominent oral/respiratory secretions, pinpoint pupils |
| Muscle disease a. Polymyositisa b. Dermatomyositisa c. Periodic paralysis d. Muscular dystrophy (oculopharyngeal muscular dystrophy, mitochondrial disorders) | Elevated serum creatine kinase in (a, b). NCS/EMG shows prominent spontaneous activity and myopathic features on EMG (c) Serum hypokalemia/hyperkalemia. History of repeated episodes with recovery in between. Respiratory, autonomic, extraocular involvement extremely rare Muscular dystrophies have normal pupillary reflexes, diplopia is rare |
aMore common etiologies.
ACE, angiotensin-converting enzyme; AChR, acetylcholine receptor; CMAP, compound muscle action potential; CNS, central nervous system; CSF, cerebrospinal fluid; EMG, electromyography; LEMS, Lambert–Eaton myasthenic syndrome; LES, Lambert–Eaton syndrome; LP, lumbar puncture; MG, myasthenia gravis; MRI, magnetic resonance imaging; NCS, nerve conduction studies; PCR, polymerase chain reaction.
Given the widespread pure motor dysfunction, three broad categories of disorders should be considered: myopathy, motor neuropathy/neuronopathy, and neuromuscular junction disease. Few myopathies produce rapidly progressive weakness and early bulbar/respiratory signs. Although the periodic paralyses may cause acute paralysis, involvement of ocular and facial muscles is rare.
The presence of sensory symptoms, prodromal viral or diarrheal illness, and an ascending pattern of weakness suggests Guillain–Barré syndrome (GBS). However, early in the course, it may be difficult to differentiate some GBS variants, such as the Miller-Fisher syndrome (MFS) or the cervical–pharyngeal–brachial variant, from botulism. Anti-GQ1b antibodies may be helpful in this clinical scenario, but results of testing may be delayed. Electrophysiological testing (showing the typical findings of demyelination) is required to rule out GBS or MFS. Tick paralysis classically has an ascending pattern resembling GBS and a careful search of the skin, especially the scalp, for tick exposure helps make the diagnosis. Poliomyelitis and West Nile infections usually cause asymmetric weakness, in association with a febrile onset and signs of meningeal irritation.
The neuromuscular junction transmission disorders – myasthenia gravis (MG) and Lambert–Eaton syndrome (LES) – resemble botulism in that they result in weakness caused by abnormal neuromuscular transmission. In addition to weakness, areflexia and autonomic involvement are common to both LES and botulism, but, in LES, cranial nerve involvement is typically not conspicuous and respiratory failure is rare. The facilitation/potentiation of muscle stretch reflexes, reported in LES, has not been observed in botulism. In MG, reflexes are typically preserved unless there is severe limb weakness. Serological studies for MG, and for LES, if positive, help make the distinction. The major distinguishing feature is that MG and LES present in a subacute to chronic manner while botulism is acute and progresses precipitously. Hypermagnesemia or the administration of other drugs adversely affecting neuromuscular transmission may cause rapid neuromuscular blockade, but is ruled out by the history.
Diagnosis of Botulism
The clinical diagnosis of botulism is supported by electrophysiological and microbiological studies. Important investigations that should be normal in botulism include: complete blood count (CBC), imaging of the brain and spinal cord, and cerebrospinal fluid (CSF) glucose, protein, and cell count.
Electrophysiological Findings in Botulism
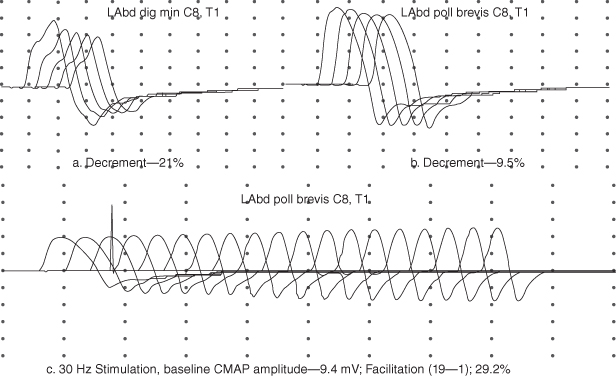
A low-amplitude compound muscle action potential (CMAP) in a clinically weak muscle is seen in up to 85% of botulism cases. However, the sensitivity of this finding may be as low as 50% when testing is limited to distal muscles routinely performed in the electromyography (EMG) laboratory. A decremental response (>10% reduction in amplitude or area) on slow repetitive nerve stimulation (2–5 Hz) is seen in both presynaptic and postsynaptic disorders of the neuromuscular junction, including botulism (Figure 17.2). Post-tetanic facilitation, after 10 s of maximal isometric exercise or with high-frequency repetitive stimulation at 30–50 Hz is seen in up to 62% of botulism cases. The degree of facilitation is modest, typically ranging from 20% to 60%, whereas facilitation in LES is typically 100–300%.
Figure 17.2. Top: repetitive nerve stimulation at 3 Hz of the left abductor digiti minimi (Abd Dig Min) and left abductor pollicis brevis (Abd Poll Brevis) muscles. Bottom: high-frequency repetitive nerve stimulation showing modest pseudofacilitation in the abductor pollicis brevis muscle.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


