Brain Tumors in the First Two Years of Life
Brain tumors manifesting in children younger than 2 years comprise a unique realm of neuro-oncology. These lesions deserve separate consideration for several reasons, all of which can profoundly impact the eventual outcome of a very young patient. Their anatomical distribution strongly favors the supratentorial compartment, whereas a predilection for the posterior fossa is seen in tumors in older children. Because of this distribution, in combination with an immature neurocognitive status, their clinical presentation is manifested not so much with neurologic findings, but rather with behavioral patterns and macrocrania of a relatively protracted and insidious duration. As a result, these tumors can achieve inordinately large dimensions before coming to medical attention and even before a correct diagnosis. Some histologic variants are so unique as to be rare in children beyond infancy. Surgical considerations that directly influence surgical morbidity include a relatively small circulating blood volume, poor thermoregulation, and incomplete skull maturity. Intraoperative adjuncts, including cortical mapping and stereotactic guidance, are rarely possible. Lastly, the therapeutic strategy needs to be tailored because of the well-recognized inverse relationship between age and potential treatment toxicity. We detail these features in this chapter, with special emphasis on the recent advances that continue to support the concept of segregating the brain tumors of very young children as a separate clinical entity.
33.1 Terminology
Previous attempts have been made to categorize young children with brain tumors according to the time of tumor genesis. Boldrey et al in 1950 proposed a definition based upon the time of symptom onset. According to this classification, children were labeled as having congenital, neonatal, or infantile brain tumors if symptoms were detected at birth, before 2 months of age, and between 2 and 12 months of age, respectively.1 Solitare and Krigman in 1964 recognized the deficiency in this strategy because of the potential discordance between tumor origin and symptom onset.2 They thus proposed that tumors be considered “definitely congenital” if symptoms were present at birth, “probably congenital” if symptoms occurred within 1 week, and “possibly congenital” when symptom onset was between 1 and 4 weeks of life. These early attempts to subcategorize and label infantile brain tumors are problematic.
In this chapter, we do not attempt to further subcategorize brain tumors in children younger than 2 years, with the exception of those tumors that are definitely congenital (i.e., symptomatic at birth). To do so presupposes an understanding of biological growth rates for varying histologic subtypes of tumors. Although it is sometimes certain that a tumor originated in utero, it is impossible to determine the time of disease onset for most children. Additionally, with any of the subclassifications outlined above, the true incidence of congenital tumors is underestimated. Spontaneous abortions and stillbirths resulting from brain tumors, estimated to account for as many as 25% of all congenital brain tumors, would not be captured in the aforementioned classification system.3–5 Furthermore, tumors that arise during embryogenesis, but do not become symptomatic until after infancy, would also be labeled incorrectly.
33.2 Epidemiology
Following a period of increasing incidence of pediatric brain tumors and of cancer generally from the 1960s through the 1990s related to improved radiographic diagnostics and reporting, the rate then stabilized.6–9 Tumors diagnosed within the first 2 years of life account for a stable proportion of childhood brain tumors, estimated at 12 to 18%.10–14 Pediatric brain tumors occur at an estimated frequency of about 4 per 100,000 person-years.15 Congenital brain tumors present at the time of birth or found in stillborns are relatively infrequent. Among all newborn and stillborn children diagnosed with cancer, brain tumors account for fewer than 5% cases, with a frequency of about 1.1 per 100,000 births.5,16 Most studies indicate a relatively equal distribution between the sexes for all tumors in aggregate, although some histologic subtypes, specifically medulloblastoma, continue to be more common in boys.4,17–20
Although they account for a small proportion of children with brain tumors, infants with brain tumors afford valuable opportunities to better understand oncogenesis. That the brain tumors encountered in infancy manifest so early, with many encountered subtypes seen exclusively in this age group, suggests a strong contributory role of germline and embryonic abnormalities, and potentially fewer contributory postnatal mutational and epigenetic events. Compared with other tumors known to originate embryonically, the relatively short interval to disease symptomatology for these tumors suggests either universal penetrance with inevitable tumorigenesis or, alternatively, a scenario of high penetrance in which required Knudson “second-hit” insults are more common, are uniquely encountered, or are exclusively carcinogenic in this time period.
Genetic predisposition has been clearly established in several congenital brain tumor syndromes. For the most part, these syndromes are transmitted as autosomal-dominant traits despite the frequent harboring of mutations in tumor suppressor genes. Thus, most tumors possess biallelic loss of function manifesting as tumorigenesis. ▶ Table 33.1 summarizes the genetic locus, the gene product, and the most common type of brain tumor associated with each syndrome. What is clear from a rapidly expanding list of congenital syndromes with known genetic bases is the fact that primary brain tumors not appearing to arise by chance alone are frequently associated with a genetic predisposition to oncogenesis or mutagenesis that readily manifests systemically. However, in the absence of a recognized congenital syndrome, it currently remains inadvisable to recommend screening of the siblings or other first-degree relatives of young children with brain tumors.
| Genetic syndrome | Gene (locus) | Gene product | CNS tumor type | Inheritance |
| Neurofibromatosis type 1 | NF1 (17q11.2) | Neurofibromin 1 | Pilocytic astrocytoma, neurofibroma, meningioma | AD |
| Neurofibromatosis type 2 | NF2 (22q12.2) | Merlin, neurofibromin 2 | Vestibular schwannoma, meningioma, ependymoma | AD |
| Tuberous sclerosis complex (Bourneville disease) | TSC1 (9q34) and TSC2 (16p13.3) | Hamartin (TSC1), tuberin (TSC2) | Subependymal giant cell astrocytoma | AD |
| von Hippel-Lindau disease | VHL (3p26-p25) | von Hippel-Lindau protein (pVHL) | Hemangioblastoma | AD |
| Retinoblastoma | RB1 (13q14.2) | Retinoblastoma protein (pRB) | Pineoblastoma | AD |
| Nevoid basal cell carcinoma syndrome (Gorlin syndrome) | PATCHED (9q22.3) | PTCH | Medulloblastoma | AD |
| Cowden disease (multiple hamartoma syndrome) | PTEN (10q23.3) | PTEN | Dysplastic gangliocytoma (Lhermitte-Duclos disease) | AD |
| Turcot or familial adenomatous polyposis syndrome | APC (5q21-q22) | APC protein | Medulloblastoma, astrocytoma | AD |
| Li-Fraumeni syndrome | TP53 (17p13.1) | p53 protein | Malignant astrocytoma | AD |
| Rhabdoid predisposition syndrome | SMARCB1, hSNF5/INI1 (22q11.2) | INI1 protein | Atypical teratoid/ rhabdoid tumor, choroid plexus carcinoma | AD |
| Abbreviations: AD, autosomal-dominant; CNS, central nervous system; PTEN, phosphatase and tensin | ||||
Epidemiologic studies have rarely identified factors that are causally related to brain tumors in children. A multitude of potential causes have been investigated, with highly variable results and commonly contradictory conclusions. What is not debated is the finding that ionizing radiation has been established as a cause of primary brain tumors.21,22 The major causes of exposure have been traced to diagnostic and therapeutic medical radiation, environmental accidents, and combat-related sources. This risk from ionizing radiation is dose- and age-dependent, with increasing doses more contributory and younger children more susceptible. Much attention and investigation have been directed toward nonionizing radiation sources (including infrared, microwave, and nearly ubiquitous radio-frequency fields, including those emanating from mobile telecommunications devices), but no conclusive evidence supports causality. Inconclusive but suggestive evidence for causality does exist for maternal and paternal dietary ingestion of cured meats, the major dietary source of N-nitroso compounds.23 The implication of parental exposure to polycyclic aromatic hydrocarbons, primarily through preconceptional smoking or occupational exposure, has historically been inconclusive, but the results of large contemporary or prospective studies suggest a relatively increased risk.24,25 Maternal exposure to viral infections during gestation has been reported to increase the odds of a central nervous system (CNS) tumor during childhood.26 Debate continues, but no strong predictive values have been found for exposure to pesticides, petrochemical products, exhaust fumes, antihistamines, and a variety of other environmental agents.
33.3 Clinical Presentation
The ability of the infant skull to compensate for volume changes, the relatively increased cerebrospinal fluid (CSF) volume within the subarachnoid spaces and cisterns, a greater degree of extracellular fluid in the parenchymal compartment, and early developmental status all affect the timing and mode of presentation of young children with brain tumors. In short, these features combine to result in a paucity of localizing neurologic signs, delayed clinical recognition, and generous tumor sizes. The most common presenting signs in this age group are thus manifestations of raised intracranial pressure, such as increasing head circumference, irritability, lethargy, emesis, and failure to thrive.14,17–20,27,28 Indeed, approximately 50 to 65% of children younger than 2 years of age with a newly diagnosed brain tumor present clinically with macrocrania with diastasis of the cranial sutures and tenseness of the fontanels. Similarly, seizures are relatively infrequent, occurring in only 10 to 15% of children at presentation. It is thus understandable that many of these children are initially diagnosed inaccurately and that the discovery of the brain tumor is quite delayed from the time of symptom onset. Most studies indicate an interval of about 8 to 12 weeks from symptom onset to diagnosis, with some reports indicating several months to years before the diagnosis was reached.
33.4 Anatomical Features
The topographic features of brain tumors in infants are distinct from those of older children. First, the relationship of tumor location to the tentorium is reversed in young children in comparison with children of older age groups. A summary of several large published series with a total of 1,252 cases indicates that in children younger than 2 years of age at diagnosis, supratentorial tumors account for 63.8% of lesions and infratentorial tumors are found in 32.4%14,17–19,27–29 (▶ Fig. 33.1). If the age group is limited to children between birth and 2 months of age at diagnosis, the distribution is further biased toward the supratentorial compartment, with 74.7% of tumors supratentorial and 17.9% located in the posterior fossa.3,32–34
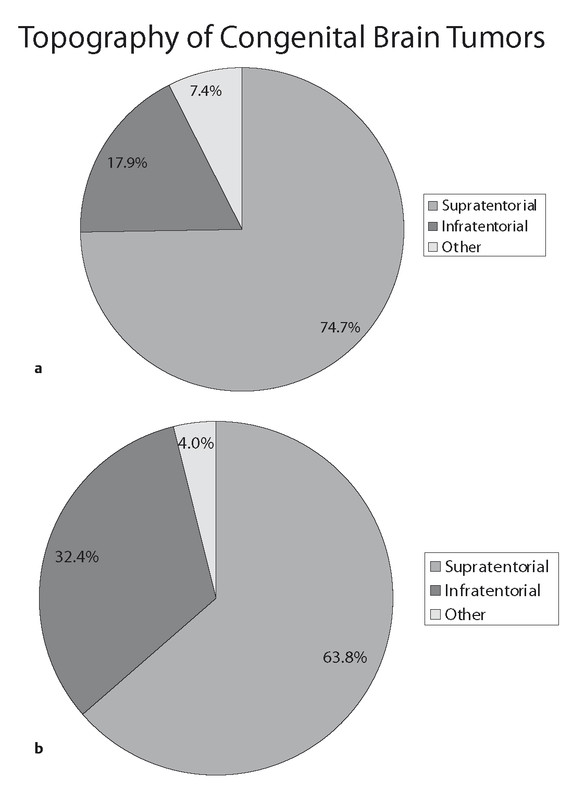
Fig. 33.1 Relative distributions of primary brain tumors in children in whom they are diagnosed before 2 years of age and in children in whom they are diagnosed at the time of birth (congenital).
Another unique feature of brain tumors in very young children is their relatively large size. These tumors frequently occupy a significant portion of the hemispheric volume. In one series from The Hospital for Sick Children in Toronto, Ontario, Canada, the average maximal diameter of tumors in children younger than 1 year of age at diagnosis was 4.6 cm, with most tumors measuring between 4 and 10 cm in largest diameter.20 Among 18 of 45 cases of congenital tumors reviewed in 1990 by Buetow et al, the tumor was noted to be “large, occupying more than one-third of the intracranial volume.”32 This finding was also noted by Tadmor et al in their 1980 treatise on children younger than 2 years of age with their comment that “…most of the tumors were extensive, involving more than one cerebral lobe.”28 These tumors can also have characteristic cystic components that are distinct from loculate hydrocephalus. Some series indicate that the tumors of up to two-thirds of very young children have macroscopic cystic components.32 An example of these particular growth features is depicted in ▶ Fig. 33.2.
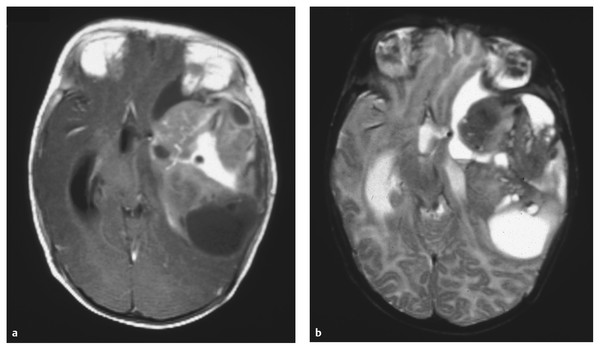
Fig. 33.2 Anaplastic astrocytoma. (a) Axial T1-weighted magnetic resonance image with contrast administration and (b) T2-weighted sequence in a 3-month-old boy who presented with failure to thrive and a divergent macrocrania. A very large mass with macroscopic cystic changes is seen in this anaplastic astrocytoma (World Health Organization grade III) of the left cerebral hemisphere. In addition to the severe mass effect, the tumor has caused ventriculomegaly, which accounts for a substantial component of the hemispheric volume.
33.5 Tumor Types
The classification of brain tumors of infancy is based on the World Health Organization (WHO) system,35 which classifies them principally according to the presumed cell of origin and degree of malignancy (grades I through IV). Although the classification system is universally used irrespective of patient age, several pathologic entities exist that remain unique to very young children and are rarely diagnosed in later childhood or adulthood. These specific tumors are found predominantly within the general categories of embryonal tumors, mixed neuronal–glial tumors, ependymal tumors, and choroid plexus tumors. Accurate diagnosis is reached through standard light microscopy, immunocytochemistry, and genetic analysis. Therefore, a detailed analysis of tumor tissue is crucial to establishing the correct diagnosis. It is clear from the experience in cooperative group studies using a central review process for pathologic interpretation that discordant diagnosis is not uncommon and potentially jeopardizes the validity of the outcome analysis.
As mentioned, the frequency of specific tumor types varies with age. The distribution of tumor types in children with a primary brain tumor who are younger than 2 years old at diagnosis is markedly different from the distribution of tumor types in older children. These relative frequencies for very young children are summarized by a review of published series cumulatively totaling 1,185 patients and are illustrated in ▶ Fig. 33.3.14,17–19,27–29
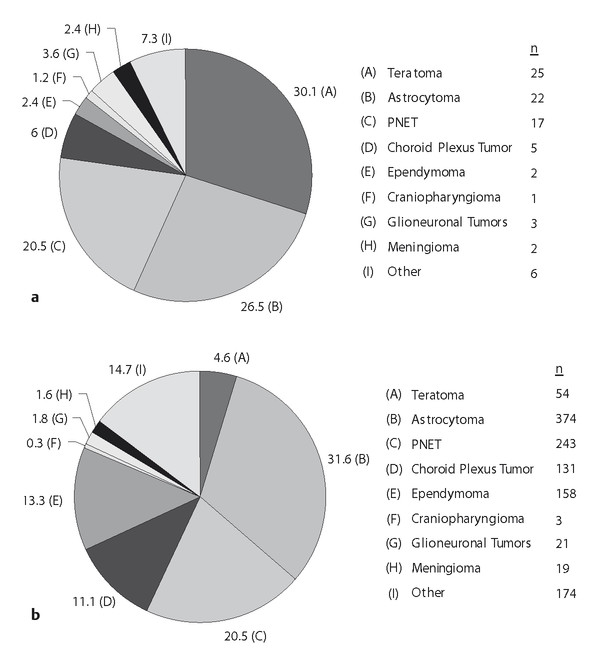
Fig. 33.3 Relative frequencies of tumor types in patients with tumors diagnosed (a) at the time of birth (congenital) or (b) within the first 2 years of life.
Notably absent from these cumulative data and the large cancer registries is the atypical teratoid/rhabdoid tumor (AT/RT). The initial description of this rare tumor, accounting for approximately 1% of all pediatric brain tumors, occurred after most of the major publications pertaining to tumors in the very young had been published.30 The majority of these tumors were most likely previously classified as primitive neuroectodermal tumor (PNET) or medulloblastoma in the period before 2000, when AT/RT was added to the WHO classification system.36 In a French registry of pediatric brain tumors, 7 of 267 tumors in patients ages 0 to 4 years were AT/RT, versus 4 of 750 in patients ages 4 to 19 years.31 In an Austrian registry of pediatric WHO grade III or IV tumors, AT/RT accounted for 13 of 75 (17.3%) high-grade tumors in patients 0 to 2 years of age, versus 6 of 236 (2.5%) high-grade tumors in children ages 2 to 14 years.37 The rate of diagnosis of AT/RT is significantly greater than that indicated in ▶ Fig. 33.3 and that indicated in large registries quantifying brain tumors in the first 2 years of life.
The major diagnostic categories of tumors in children younger than 2 years of age are described below. When appropriate, the reader is referred to the corresponding chapter for that particular tumor. Here, we highlight the main features of each of the major histologic subtypes that are unique to infants and provide a more thorough assessment of the tumors that are particular to very young children and are seldom diagnosed beyond infancy.
33.6 Emerging Developmental Insights into Tumorigenesis
Oncogenesis has classically been understood to fundamentally represent the dysregulation of processes controlling cellular proliferation, longevity, and respect for surrounding tissues. A growing body of scholarship has established that co-opted processes of normal development frequently contribute to oncogenesis; indeed, in many tumor types, cancer stem cells embodying populations of aggressive cells capable of initiating or repopulating tumors harbor features of prematurity. Given their young age at presentation, it is therefore unsurprising that putative tumor cells of origin intimately associated with developmental progenitors have also been identified among CNS tumors of infancy.
The prototypical example of this is medulloblastoma of the cerebellum, a structure particularly susceptible to tumorigenesis because it undergoes a significant proportion of its development postnatally. Medulloblastoma comprises at least four subgroups that are biologically distinct on the basis of molecular signature, clinical behavior, and response to therapy; these include the sonic hedgehog (SHH, correlated with the desmoplastic histologic variant), wingless (Wnt), and other “non-Wnt/non-SHH” genetic subtypes.38 SHH-subgroup medulloblastoma, the most common subtype encountered in infants, has been shown to arise from cerebellar granule neuron progenitor cells. During normal development, granule neuron progenitor cells undergo massive expansion before cell cycle arrest and terminal differentiation in response to decreasing paracrine SHH as they migrate beyond SHH-secreting Purkinje cells en route to the internal germinal layer, where their quiescent progeny reside as granule neurons. In disease, this subgroup exhibits aberrant signaling of SHH, associated pathways including Notch, and downstream effectors of proliferation in addition to other oncogenic abnormalities.39 Other cerebellar progenitors may similarly be responsible for medulloblastoma tumorigenesis, with recent evidence that those originating in the upper and lower rhombic lips may provide tumor cells of origin for some Wnt-subtype tumors.40 These insights have already potentially yielded therapeutic targets such as hedgehog inhibitors, and advances in developmental and tumor biology may accelerate future prognostic and therapeutic progress.41
33.7 Primitive Neuroectodermal Tumors
33.7.1 Medulloblastoma
Medulloblastoma is the most common brain tumor in infants, accounting for nearly 50% of newly diagnosed brain tumors in children younger than 2 years of age. Of all medulloblastomas diagnosed in children, 25 to 35% occur in children younger than 3 years of age.42 As expected, children with these tumors primarily present with symptoms of intracranial hypertension secondary to hydrocephalus resulting from fourth ventricular obstruction (▶ Fig. 33.4). Medulloblastomas tend to carry a worse prognosis in children younger than 2 to 3 years old. The cause of the unfavorable prognosis in this age range is speculated to result from the avoidance of radiation therapy, although some suggestion has been made that medulloblastoma in very young children may have a more aggressive biological behavior. Furthermore, leptomeningeal dissemination has been reported to be present in 27 to 43% of younger children, although this higher rate of metastatic spread is not found in all retrospective series.43–45
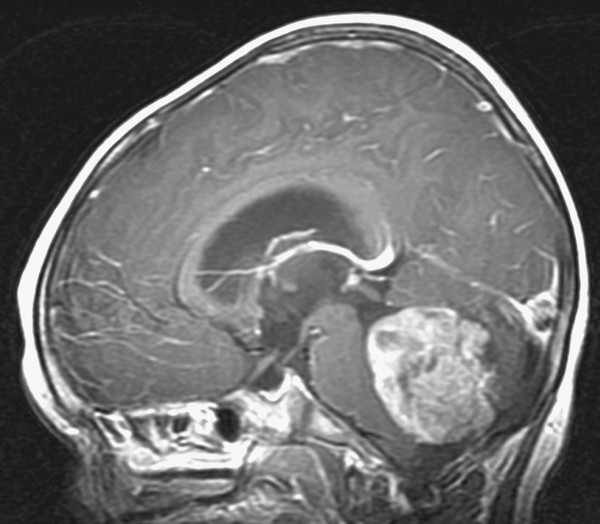
Fig. 33.4 Medulloblastoma. Sagittal magnetic resonance image following the administration of contrast in a 2-year-old boy presenting with macrocrania and intermittent morning vomiting. The fourth ventricular medulloblastoma and resulting noncommunicating hydrocephalus are easily seen on this preoperative image.
The histopathologic grading of medulloblastoma indicates that some prognostic significance is achieved by subclassifying these tumors, although commonly used treatment regimens do not currently stratify patients on the basis of pathologic interpretation. The 2000 WHO classification recognized four separate varieties of medulloblastoma: classic medulloblastoma, desmoplastic medulloblastoma, large cell/anaplastic medulloblastoma (LC/A MB), and medulloblastoma with extensive nodularity (MBEN).46 (For a detailed discussion of medulloblastoma, the reader is referred to Chapter ▶ 40.) The latter entity, MBEN, has only recently been defined and warrants separate consideration, given its predilection for very young children. MBEN, although rare and accounting for fewer than 5% of medulloblastomas, occurs most often in children younger than 3 years of age. This tumor, characterized by a cellular pattern of extensive nodular growth with strong neuronal differentiation, was formerly termed cerebellar neuroblastoma (▶ Fig. 33.5). This subtype of medulloblastoma has been shown to behave in a more indolent fashion; it metastasizes less frequently and carries a better prognosis than other medulloblastoma subtypes.47–50However, given the challenges encountered in treating very young children and their resultant poor outcome, children younger than 3 years of age with medulloblastoma, irrespective of the extent of disease or presence of dissemination, are generally categorized as being at high risk.
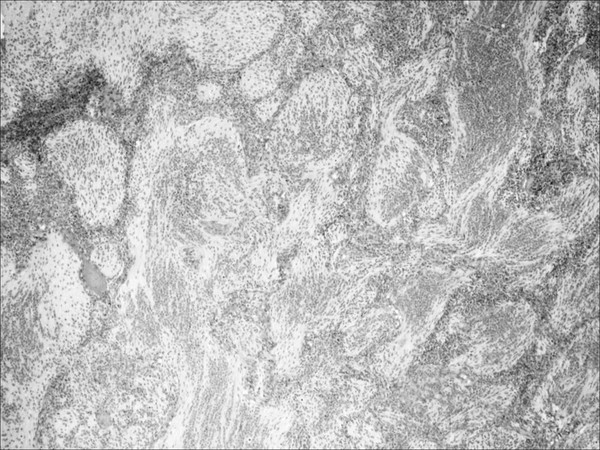
Fig. 33.5 Medulloblastoma. Low-power hematoxylin and eosin (H&E) stain of a medulloblastoma with extensive nodularity. This variant is characterized by a histologic phenotype having two major cytoarchitectural profiles. There are reticulin-free, synaptophysin-positive zones composed of well-differentiated neurocytes. These nodules are surrounded by reticulin-rich collagen regions with a subpopulation of poorly differentiated cells that do not stain for neuronal markers.
The results of cooperative group studies show that the survival rates of very young children are significantly reduced compared with those of older children. The recent trend in treatment has focused on the attempt to avoid, delay, or reduce irradiation with chemotherapeutic supplementation. In a study of 12 children with medulloblastoma diagnosed before 3 years of age at MD Anderson Cancer Center in Houston, Texas, 8 children (67%) treated with chemotherapy alone had a median survival rate of 10.6 years.51 In 1993, the Pediatric Oncology Group (POG) reported a 5-year progression-free survival (PFS) rate of 32% and an overall survival (OS) rate of 40%.52 This study employed delayed radiation therapy (see below) and found complete surgical resection to be the strongest positive predictor. The Children’s Cancer Group (CCG) employed “eight drugs in 1 day” for 46 children younger than 18 months and reported a 3-year PFS rate of 22%.43 Subsequent studies by the CCG and Société Française d’Oncologie Pédiatrique (SFOP) employing conventional chemotherapy and reserving adjuvant radiotherapy for recurrent disease or as salvage treatment have borne similar outcome analyses for very young children with medulloblastoma: an expected approximately 30% PFS and approximately 40% OS at 5 years.53,54 Recent data published from the German HIT-SKK87 registry noted approximately 50% and 60% PFS and OS at 10 years, respectively, in children younger than 3 years of age who were without either postoperative residual or macroscopic metastases at diagnosis and were treated with upfront adjuvant chemotherapy and craniospinal irradiation after age 3.55
Other treatment regimens, including intensified chemotherapy and intrathecal routes of administration, have also been reported (see below). Of particular note, a recent study that utilized an intensive chemotherapeutic approach with intrathecal supplementation in children younger than 3 years of age reported favorable results.42
Thus, children with medulloblastoma who are younger than 3 years of age at diagnosis are at a substantial disadvantage compared with older children in respect to overall outcome. The extent of surgical resection and degree of metastatic disease appear to influence outcome. Intensive chemotherapy and intrathecal routes are being used with increasing success to eliminate or reduce radiation exposure. Although histologic subtypes have not been effectively used to date in the prognosis or treatment of medulloblastoma, the recent description of molecular subtyping (including aberrant SHH signaling, most frequently seen in infants) and increasing knowledge of the cellular programs gone awry in the clinical syndrome will doubtless influence treatment goals, suggest avenues for drug design and delivery, and assist in tailoring therapy to an infant’s individual disease.56
33.7.2 Supratentorial Primitive Neuroectodermal Tumors
Long recognized as the hemispheric equivalent of the medulloblastoma, the supratentorial primitive neuroectodermal tumor (sPNET) clearly has a worse outcome than those morphologically similar tumors of the cerebellum. These highly malignant neoplasms account for only 2.5 to 7% of childhood brain tumors, but they predominate in the very young.57 In the infant population, these tumors can achieve very large dimensions and thus present an onerous challenge for surgical removal. In the 1995 summary of sPNETs from the CCG, 40% of the tumors had a maximal diameter of more than 6 cm; total resection was accomplished in only 37% of patients.58 The POG review of sPNETs also reported the large dimensions of these tumors, with an average volume of 153.5 cm3.59 In addition, these tumors frequently were found to involve more than one cerebral compartment, thus precluding the possibility of aggressive resection. As reported by the COG, the 5-year OS rate of 34% and PFS rate of 31% were statistically worse in children younger than 3 years at diagnosis.58 In fact, that group showed a substantial reduction in the 3-year PFS rate from 53% for children younger than 3 years to 25% for children between the ages of 1.5 and 2 years old.60
Some improvement in outcome has been purported with the use of dose intensification or high-dose chemotherapy and autologous bone marrow transplantation. Memorial Sloan-Kettering Cancer Center (MSKCC) reported a 2-year event-free survival (EFS) rate of 43% for 14 patients.61 In that particular series, however, 50% of the patients were older than 30 months of age at diagnosis, pineoblastoma was grouped together with sPNET, and there was a 6% treatment-related mortality rate. Among infants enrolled in the German HIT-SKK87 and SKK92 registries (ages 37 months or younger), the benefit of radiation therapy was of a magnitude (28.6% OS and 24.1% PFS among those treated with radiotherapy vs. 6.7% OS and PFS at 3 years among those with adjuvant chemotherapy alone) that prompted the authors to recommend that radiation be delayed by no longer than 6 months; however, this approach remains uncommon.62
33.7.3 Pineoblastoma
PNETs of the pineal region, or pineoblastomas, are malignant embryonal tumors that arise from the pineal parenchymal cells (▶ Fig. 33.6). For the most part, pineoblastomas are histologically indistinguishable from PNETs in other locations. Their behavior has been difficult to define, given the tendency to group this entity together with other malignant neoplasms of the pineal region, the historical therapeutic approach of using empiric radiation therapy, and the inaccurate assignment of the tumors to other histologic categories.
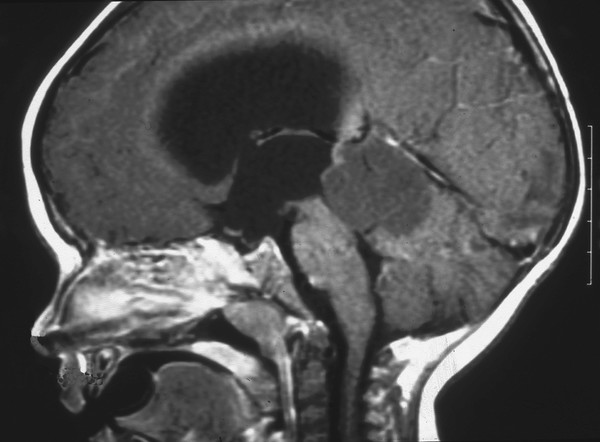
Fig. 33.6 Pineobladtoma. Sagittal magnetic resonance image following contrast administration in a 1-year-old boy who presented with macrocrania and vomiting. The lesion in the pineal region has obliterated the normal anatomical structures, causing obstruction at the level of the aqueduct and severe hydrocephalus. Histologic samples following total resection were indicative of a pineoblastoma (primitive neuroectodermal tumor of the pineal region).
A very young child with a pineoblastoma has a significantly poor prognosis. Although justified, withholding irradiation is the most probable reason. The 1995 report by the CCG indicated widely disparate outcomes for children with pineoblastoma depending upon age at the time of diagnosis. The 3-year EFS rate for children younger than 18 months of age treated with chemotherapy alone was 0%, compared with a 3-year EFS rate of 61% for older children treated with combined radiation therapy and chemotherapy (▶ Table 33.2).63 In the infant population, the median time to disease recurrence was 4 months, and the median time to death was 10 months. The POG reported similarly dismal results in 11 infants with pineoblastoma when an approach of postoperative chemotherapy with the intent to delay radiation therapy was used. All patients failed chemotherapy, no patients had a response of their leptomeningeal disease, no children were salvaged with radiation therapy, and all children died of disease between 4 and 13 months from the time of diagnosis.64
The use of high-dose chemotherapy with autologous stem cell rescue is being considered as the primary approach in young children with pineoblastoma, given the extremely poor rates of response to conventional chemotherapy without irradiation. No large cooperative study using such an approach for infants with pineoblastoma has been completed, but pilot study data indicate its feasibility. Of 13 patients with pineoblastoma recently treated at Duke University with high-dose chemotherapy and autologous stem cell rescue, two were younger than 1 year at the time of treatment.65 These two patients, who had localized disease and underwent a complete surgical removal, had a continuous complete remission of 35 and 125 months’ duration after treatment.
33.7.4 Trilateral Retinoblastoma
Trilateral retinoblastoma is a unique and particular tumor syndrome of infancy. This entity is defined as the composite of a previous history of bilateral retinoblastoma with a subsequent diagnosis of an intracranial PNET (▶ Fig. 33.7). Although subsequent intracranial PNET has been recognized in children with unilateral retinoblastoma, this is the rare exception. The overall incidence of an intracranial PNET, typically a pineoblastoma, in the presence of bilateral retinoblastoma is 2 to 11%.66 The mean age of a child at the time of presentation with an intracranial tumor is about 24 months. In most reports, trilateral retinoblastoma has been uniformly fatal, with a mean length of survival after discovery of the intracranial tumor of 1.3 months for untreated patients and 9.7 months for treated patients.67 The very poor outlook is partly due to the fact that up to 25% of patients have disseminated disease when the intracranial tumor is discovered. There is some evidence to support the use of neoadjuvant intravenous chemotherapy (chemoreduction) in children with bilateral retinoblastoma or a family history of retinoblastoma to reduce the incidence of trilateral retinoblastoma (▶ Table 33.2).68
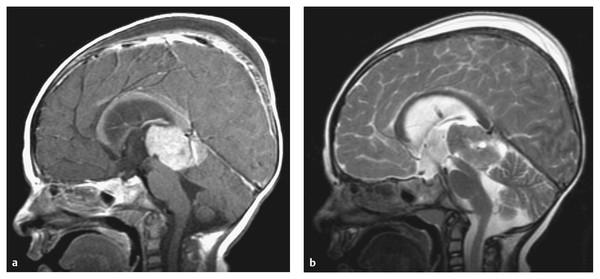
Fig. 33.7 Trilateral retinoblastoma. (a) Sagittal T1-weighted magnetic resonance image with contrast and (b) the corresponding T2-weighted sequence of an 11-month old girl previously treated for congenital bilateral retinoblastoma. The images were obtained after an endoscopic third ventriculostomy and tumor biopsy (note the turbulent flow pattern at the site of the third ventriculostomy). This child with trilateral retinoblastoma underwent total resection of a pineoblastoma following induction chemotherapy.
| Author (year) | Patients | Chemotherapy regimen | Response |
| St. Clair et al (1991–1992) | 4 | Etoposide/ifosfamide/ carboplatin | 4 PR |
| Packer et al (1992)94 | 1 | Cyclophosphamide/vincristine | 1 PD |
| Allen et al (1992)99 | 3 | Etoposide/cisplatin | 2 PR, 1 SD |
| Arico et al (1994) | 1 | Teniposide/procarbazine/intrathecal methotrexate/vincristine/ lomustine | 1 SD |
| Duffner et al (1995) | 4 | Vincristine/cyclophosphamide | 2 PR, 2 SD |
| Razzaq and Cohen (1997)97 | 1 | Etoposide/ifosfamide/ carboplatin | 1 PR |
| Total | 14 | 9 PR, 4 SD, 1 PD | |
| Abbreviations: PD, progressive disease; PR, partial response; SD, stable disease. aIncludes only patients with measurable disease following subtotal surgical resection or biopsy. | |||
33.8 Ependymoma
Ependymoma of childhood is a rare tumor with an incidence of 2.1 to 2.5 per 1 million children younger than 15 years.69 (The reader is referred to Chapter ▶ 41 for a complete discussion of ependymoma of childhood.) With a median age at onset in most large series between 36 and 52 months, the infant population represents a disproportionate fraction of children affected.70,71 In children younger than 2 years, the majority of these tumors are fourth ventricular in location. Ependymomas in infants are found less frequently in the supratentorial compartment, where they may be entirely intraparenchymal and have no relationship to the ventricular compartment. Those tumors arising within the cerebral hemispheres can attain voluminous sizes due to their relatively indolent growth rates (▶ Fig. 33.8).
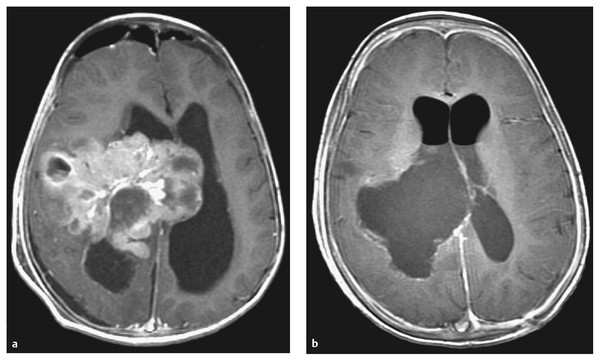
Fig. 33.8 Ependyoma. (a) Preoperative and (b) postoperative axial magnetic resonance images following contrast administration in a 14-month-old boy with a supratentorial ependymoma. The tumor occupies a significant portion of the right cerebral hemisphere. The child underwent total excision of an anaplastic ependymoma at second-look surgery following adjunctive preoperative chemotherapy.
A young age at the time of diagnosis has been repeatedly shown to negatively affect outcome in children with intracranial ependymoma. Proposed reasons include a lower rate of complete resection, the avoidance of radiation therapy, and biologically aggressive behavior.70,72 The outcome for infants treated for ependymoma was a 3-year PFS rate of approximately 26% as assessed by the CCG.43 Although whether pathology is predictive of outcome is controversial, the CCG study enrolled only patients with a malignant or anaplastic histology. The POG evaluated 48 children younger than 3 years with ependymoma (classic and anaplastic varieties) and found a radiologic response rate of 48% with a treatment approach that used neoadjuvant chemotherapy with the intent of delaying or completely avoiding radiation therapy. The extent of resection was found to be the most important predictor of outcome, with 5-year OS rates of 66% and 25% for those children undergoing gross total excision and subtotal excision, respectively. The study also established that the disease control rates, which are similar shortly after therapy, diverge with longer follow-up between patients younger than 1 year and those between 2 and 3 years of age at the time of treatment. Thus, at 5 years, the younger patients had a 26% survival rate, whereas the older children had a survival rate of 63%. The conclusion drawn from these important observations was that a delay of radiation of more than 1 year adversely affects survival.
As is true in children of all ages, the principal site of recurrence is usually at the primary site of disease, with a minority of children having disseminated disease at the time of progression. The pattern of recurrence and poor control rates have led to the use of highly conformal radiation therapy as a therapeutic adjunct. This approach has been used in 88 patients whose mean age was 2.85 years, with 15 patients younger than 18 months at the time of treatment, at St. Jude Children’s Research Hospital in Memphis, Tennessee.73 With a median follow-up of over 38 months, the 3-year PFS rate was 75%. In interpreting these data, it is important to bear in mind that 84% of the patients had confirmed gross total excision of their tumor before irradiation.
Because of the wide recognition that the extent of surgical excision positively affects outcome, other strategies, including surgical re-exploration (so-called second-look surgery), have been advocated for residual ependymoma. This approach has been used in small, single-institution series with initial success toward achieving gross total excision.74,75
33.9 Astrocytoma
A detailed account of astrocytomas in children can be found in Chapters ▶ 42 and ▶ 43. However, several characteristic features of astrocytomas in very young children warrant separate consideration.
33.9.1 Optic Pathway Gliomas (Chiasmatic/Hypothalamic Gliomas)
Gliomas of the optic pathway (chiasmatic/hypothalamic gliomas) account for nearly 20% of all intracranial tumors in children younger than 2 years.76 These children can present with several manifestations. Alterations in vision usually manifest as nystagmus, spasmus nutans, or poor fixation. Raised intracranial pressure from noncommunicating hydrocephalus is most frequently identified by progressive macrocrania. The diencephalic syndrome of emaciation, or Russell syndrome, is quite specific for a hypothalamic glioma during infancy; it is characterized by failure to thrive, normal axial growth, and increased growth hormone levels.77 The most frequent manifestations of alterations in endocrine function are precocious puberty and growth hormone deficiency. Although neurofibromatosis type 1 (NF1) is present in roughly half of all children with chiasmatic/hypothalamic glioma, most children younger than 2 years in whom this tumor is diagnosed do not have NF1.78 Although the less frequent association with NF1 may in part be responsible for the poorer prognosis of children younger than 2 years of age, the tumor may have a more aggressive biological behavior in very young children, and there is a reluctance to utilize radiation therapy. The current treatment strategy includes chemotherapy only in the presence of symptom progression or radiographic changes. With such an approach, the 5-year PFS rate for children younger than 3 years of age at diagnosis has been reported at 63%.78
33.9.2 Pilomyxoid Astrocytoma
A recently described variant of hypothalamic/optic pathway pilocytic astrocytoma, pilomyxoid astrocytoma has been recognized to have a predilection for younger children; the mean age at onset is 18 months.79–81 This subtype of pilocytic astrocytoma is identified on the basis of the histologic phenotype, but the diagnosis carries a different set of clinical features. In addition to causing symptom onset at a younger age, these tumors exhibit a more aggressive behavior, a higher rate of local recurrence, a substantial rate of CSF dissemination, and a shorter PFS (mean, 26 months) and OS (mean, 63 months).79 The histologic identification is based on a monomorphous rather than a biphasic pattern of cells on a myxoid background. The tumor is notably absent in Rosenthal fibers and has rare eosinophilic granular bodies, features quite commonly found in the pilocytic counterpart. The recognition that pilomyxoid astrocytoma occurs predominantly in the infant age group may partly explain the well-known association between poor prognosis and young age in children with hypothalamic/optic pathway astrocytomas. At the current time, the treatment of pilomyxoid astrocytoma is analogous to that of pilocytic astrocytoma, but with better classification of this subtype, the therapeutic approach may change.
33.9.3 Supratentorial High-Grade Astrocytoma
The malignant astrocytomas (WHO grades III and IV) that occur in young children, although much less frequent, appear to be unique compared with similar histologic tumors in older children and adults. The overall outcome in children with malignant astrocytoma is more favorable in most series. However, infants with malignant astrocytoma do not possess the same prognostic advantage realized in older children. Infants with malignant astrocytoma present a treatment dilemma, not unlike infants with other supratentorial malignant tumors, owing to the large size and hemorrhagic potential of the tumor and to treatment limitations caused by adverse events related to therapy. The CCG experience concerning the outcome of 39 children younger than 24 months of age treated with neoadjuvant chemotherapy indicated an overall response rate of 24%.43 The most significant variable affecting outcome was tumor pathology, with 3-year EFS rates of 44% and 0% for anaplastic astrocytomas and glioblastomas, respectively.
Some molecular analytic data indicate that malignant astrocytomas in infants are biologically distinct. Mutations in the tumor suppressor gene TP53 appear to be distinctly less frequent in tumors from children younger than 3 years of age than in those from older children and adults.82 Given that overexpression of p53 has been correlated with an unfavorable prognosis for children with malignant gliomas, this reduced incidence of mutagenesis may indicate a better response rate to particular therapies.83
33.10 Choroid Plexus Tumors
Choroid plexus tumors are truly unique tumors of very young children, with the majority of patients younger than 3 years of age at the time of diagnosis.84 Choroid plexus tumors account for approximately 0.5% of all intracranial tumors but nearly 12% of all brain tumors in children younger than 2 years of age. Hydrocephalus invariably exists in children with choroid plexus tumors. The mechanism is probably multifactorial, but this entity represents the only known common cause of CSF overproduction resulting in hydrocephalus (▶ Fig. 33.9). After complete tumor removal, treatment of hydrocephalus may still be needed, given the likely association with intraventricular hemorrhage or inflammatory reactions resulting from surgery and mechanical distortion of the intraventricular CSF pathways. The rate of resulting need for CSF diversion by way of shunting has been reported to be as high as 75%.85 Subdural effusions tend to require further management nearly as frequently as hydrocephalus. The large size of the tumor, degree of ventriculomegaly, and extended duration of the surgical cases all probably contribute toward this postoperative management issue. Closure of the corticectomy site with fibrin sealant following tumor removal has shown some success in isolating the intraventricular compartment from the subarachnoid space.
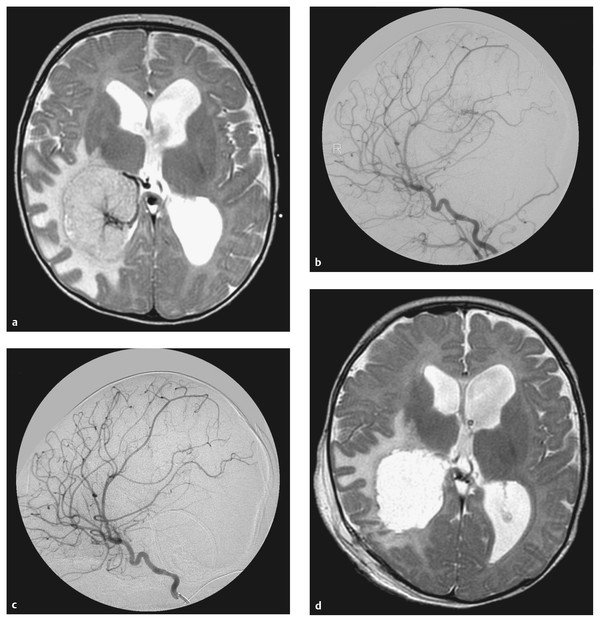
Fig. 33.9 Choroid plexus papilloma. Representative magnetic resonance images from a 4-month-old boy who was evaluated for increasing head circumference. (a) An axial T2-weighted image revealed a large left intraventricular mass and ventriculomegaly. Given the expected tumor vascularity, (b) an angiogram was obtained that confirmed the presence of a very rich arterial supply. The lateral projection of the right internal carotid artery depicts the tumor blush principally from the right anterior choroidal artery. (c) The postembolization angiogram demonstrates a reduction in vascular blush. (d) The postoperative axial T2-weighted image confirms that a gross total excision of the tumor was accomplished.
These tumors are some of the most surgically challenging lesions in infants owing to their large size, invasiveness, and hypervascular nature, with intraoperative mortality not infrequently reported resulting from blood loss.87 The surgical approach needs to be tailored such that the vascular pedicle or tributaries are identified early and controlled before the tumor is removed. Depending on the site within the lateral ventricle, a high parietal approach offers early recognition of the medial posterior choroidal vessels, whereas an inferior temporal approach offers early control of arterial feeders from the anterior choroidal artery through the choroidal fissure in the temporal horn of the lateral ventricle. Some attempts to reduce tumor vascularity preoperatively with endovascular embolization have been reported.86–89 These attempts have historically been limited, given the technical challenges associated particularly with cannulating the medial posterior choroidal vessels; however, when possible, the benefits are apparent and may contribute toward a simpler resection. One recent report even suggests complete remission of a presumed choroid plexus papilloma by embolization alone at 16-month follow-up.90 An example of such an approach is shown in ▶ Fig. 33.9.
Choroid plexus papilloma (WHO grade I) occurs about twice as often as choroid plexus carcinoma.87 The differentiation of these tumors is based on careful interpretation of the pathologic specimen (▶ Fig. 33.10). Contrary to the predominantly fourth ventricular location seen in adults, the lateral ventricle is the most common location for choroid plexus papilloma in children. These tumors are treated with primary resection, which in contemporary series is achieved in as many as 96% of cases.87 With complete excision, the 5-year survival rate has been reported to be as high as 100%. The atypical choroid plexus papilloma (WHO grade II) is an entity of intermediate malignancy and prognosis relative to papilloma and carcinoma of the choroid plexus.91
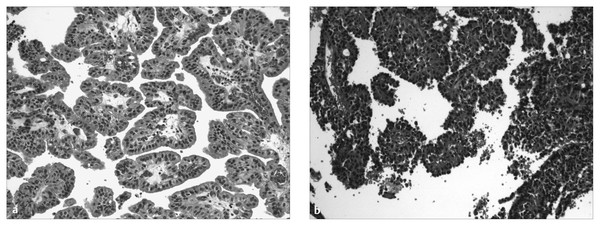
Fig. 33.10 Choroid plexus tumors. Histologic samples from (a) a choroid plexus papilloma and (b) a choroid plexus carcinoma. Both recapitulate a papillary cytoarchitecture, but the carcinoma shows foci of solid growth and prominent cytologic atypia.
Choroid plexus carcinoma (WHO grade III) is a rare disease, accounting for 1 to 4% of all brain tumors in children. This tumor differs from its more common counterpart, choroid plexus papilloma, in that it most frequently arises within the lateral ventricle, presents during infancy, is frequently invasive, and has a relatively poor prognosis. The 5-year OS rate ranges from 40 to 50%.92,93 Treatment modalities have ranged from surgery alone to surgery with postoperative radiation therapy and chemotherapy. Most reports suggest that the prognosis is improved with complete surgical resection87,92–95 and the use of adjuvant chemotherapy.87,93–100Although no consensus exists about the value of chemotherapy after total excision, published response rates with measurable disease certainly indicate that there may be a benefit in this context. Complete surgical resection is hampered by the typically large size and vascularity of these tumors in young children with a proportionally small circulating blood volume. In one contemporary study, the rate of surgically related mortality due to complications of excessive hemorrhage was as high as 30%.87 Irradiation, whose role is supported by some previous evidence, has fallen out of favor, given the typically young age of afflicted children and the known detrimental sequelae of irradiation during infancy.
Loss of function of TP53, implicated in a variety of tumors, as previously mentioned, and responsible for the Li-Fraumeni syndrome, has recently been shown to be associated with higher grades and poorer outcomes in the approximately 50% of sporadic and syndromic choroid plexus tumors in which it is found, compared with the intriguingly large fraction of these tumors that exhibit other alterations in this gene.101 While mechanistic studies are ongoing, screening for Li-Fraumeni syndrome and molecular analysis of somatic tumor tissue may play an important prognostic role in the future treatment of this disease.102,103
33.11 Atypical Teratoid/Rhabdoid Tumor
Since the earliest description of atypical teratoid/rhabdoid tumor (AT/RT) as a separate entity, better clinical descriptions have become available, and the tumor has undergone molecular characterization as well.36 AT/RT frequently occurs in children younger than 2 years of age; the mean age at diagnosis is 17 months.36,104,105 The tumor has a predilection for the posterior fossa and pineal region; however, it also occurs with some frequency in the supratentorial compartment. In fact, the tumor occurs about twice as frequently in the infratentorial compartment as in the supratentorial location. Surgical resection is challenging owing to the hypervascular nature of this tumor, and the difficulty is compounded by the young age and relatively small circulating blood volume of the patients.
The histologic appearance reflects the malignant nature of the tumor, evidenced by a compact and highly cellular lesion with widely pleomorphic features, mitotic figures, and areas of necrosis. The small, compact cytoarchitecture is reminiscent of medulloblastoma, and it is likely that many of these tumors are incorrectly diagnosed as such36 (▶ Fig. 33.11). A population of large, pale cells with “rhabdoid” features is responsible for the current nomenclature. The embryonic nature of the tumor is confirmed by its immunophenotypic diversity, which indicates mesenchymal and epithelial differentiation (vimentin, glial fibrillary acidic protein, epithelial membrane antigen, cytokeratins, synaptophysin, chromogranin, and smooth muscle actin).106 Chromosomal and genetic analysis confirms that this tumor differs from other highly malignant embryonal tumors. Cytogenetic analysis yields the frequent loss of chromosome 22, with involvement of the chromatin-remodeling tumor suppressor gene INI1/hSNF5 detectable in a majority of cases.107–111
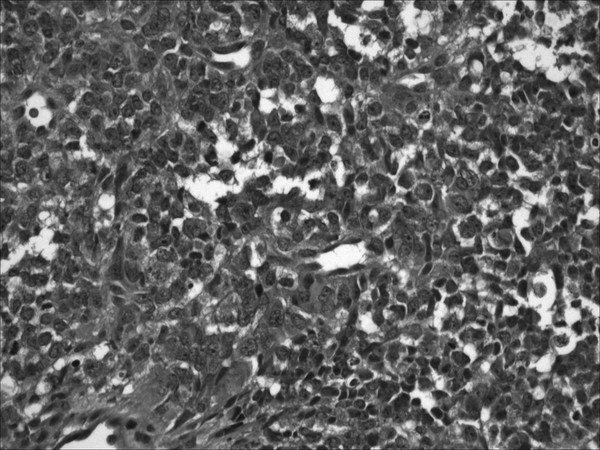
Fig. 33.11 AT/RT. Low-power hematoxylin and eosin (H&E) stain of an atypical teratoid/rhabdoid tumor. Large cells contain moderate amounts of pink cytoplasm, hyperchromatic nuclei, and prominent nucleoli. The compact nature of the tumor with a notable frequency of mitotic figures is reminiscent of a medulloblastoma. Note the large vacuoles, which produce a “starry sky” pattern.









