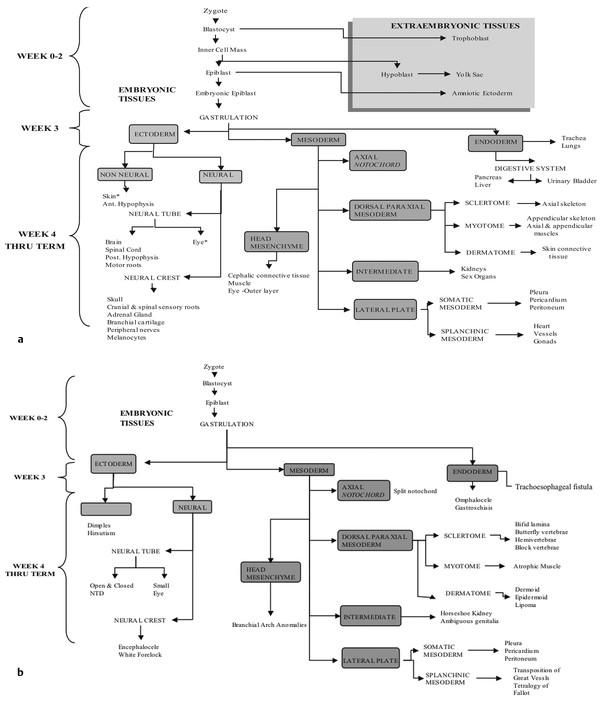
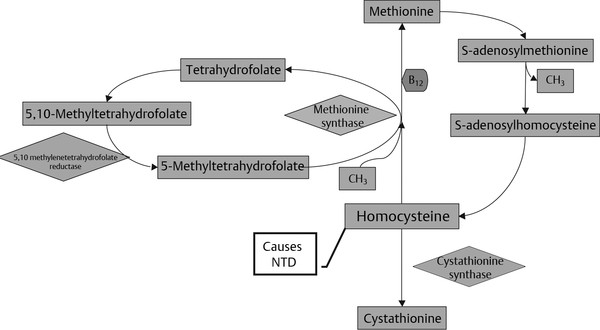
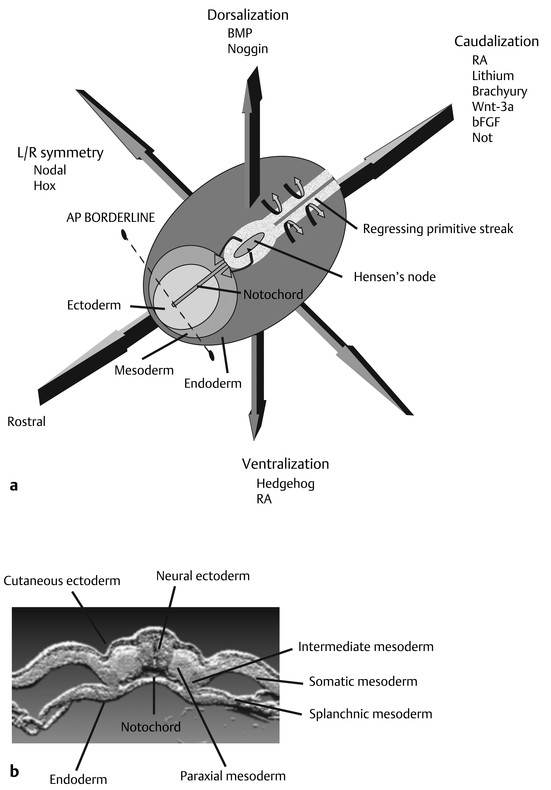
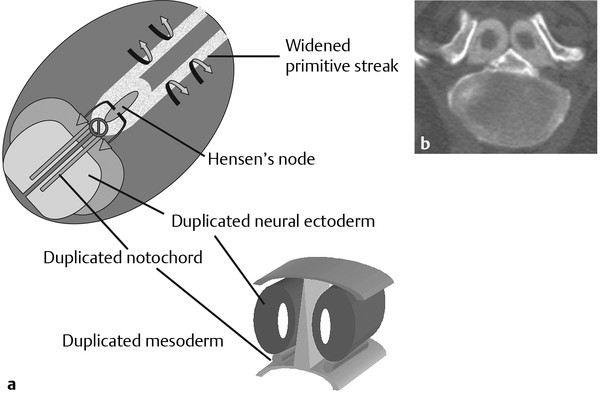
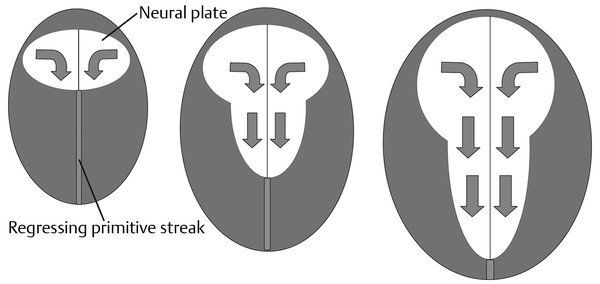
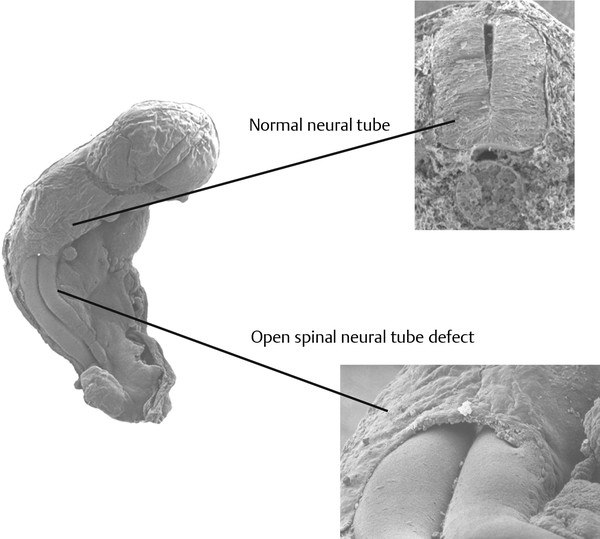
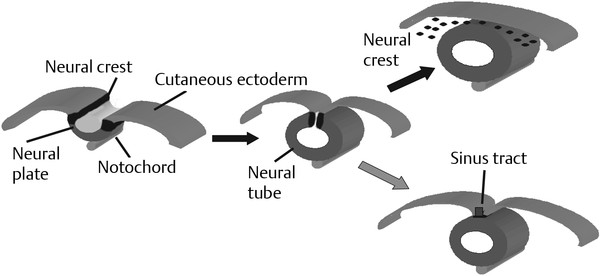
Normal and Abnormal Development of the Nervous System Understanding normal development and the consequences of abnormal early embryogenesis remains vital for the surgeon caring for children with congenital nervous system anomalies. In this chapter, we discuss nervous system defects as they relate to particular embryologic events and the mechanisms causing the malformations. However, a complete description of the development of the human nervous system is a Herculean task beyond the scope of this chapter; therefore, we refer the extensive details of neuroembryology to expert texts1,2 and the Web site (http://www.med.unc.edu/embryo_images) for excellent images. It is always important to bear in mind that the central nervous system (CNS) develops in concert with other organ systems (▶ Fig. 2.1a). In fact, the development of other organ systems is often instructed by proper CNS development and vice versa. As such, maldevelopment of the CNS is typically coupled to malformations of another organ system (▶ Fig. 2.1b). From a modern embryologic perspective, the mechanisms causing associated malformations must be studied interdependently. This approach allows an examination of the molecular signaling, tissue interactions, cellular differentiation, and cellular orientation that define organogenesis. Fig. 2.1 Developmental sources and timeline of embryonic tissues. (a) Normal developmental structures. (b) Abnormal developmental structures. The embryopathy for many defects has not been tested experimentally. We are, therefore, left with many speculative and observational theories to explain the embryogenesis of human malformations. We do not exhaustively review all theories here but give the most recent view on embryopathy based on best experimental data. Only when relevant to the embryology does a broader discussion of multiple theories take place. Secondarily acquired CNS pathology, such as hydrocephalus or syringomyelia, is not discussed. The evaluation of how molecules effect development and disease can be divided into three categories: genomics, the study of DNA and RNA structure and regulation; proteomics, the study of the control and regulation of protein products within the cell and tissue matrix; and metabolomics, the exploration of pathways metabolites use to govern cellular function. Diseases occur as a disturbance of one or more of these areas. Future diagnostics and treatments will be based on the understanding and manipulation of these molecular substrates. Congenital defects of the nervous system do not fit into simple genetic syndromes but more often embody complex genetic disorders. For example, many regulatory, structural, and enzymatic genes have been implicated in neural tube defects (NTDs).3 Epidemiologic studies of potential environmental risk factors for NTDs have a relatively long history and have identified several factors (e.g., valproate, maternal diabetes, Agent Orange) that appear to be related to NTD risk. In contrast, the study of genetic risk factors for NTD has a relative short history. Although traditional approaches to genetic linkage (e.g., LOD [logarithm of the odds] score and affected relative pair analyses) have been applied to the study of other conditions since the 1980s, the data required by such approaches (i.e., DNA from multiple affected individuals within families) are largely unavailable for NTDs. Insights from the human genome project4,5 have suggested that the genome represents a complex biochemical machine functioning in three-dimensional space with three distinct and dynamic interacting parts. DNA coding regions, only 2% of the genome, are responsible for blueprinting protein structure, the repository of heritable traits. “RNA only” coding DNA gives rise to active RNAs capable of altering the behavior of normal genes. Consequently, studies designed to identify genes that influence the risk for NTDs did not become feasible until the 1990s, when the Human Genome Project began to provide new information regarding the variability within the human genome. Such advances in our understanding of the human genome have provided new opportunities for studying the genetic contribution to conditions like NTDs (e.g., case–control and family-based genetic association studies of non-Mendelian conditions). Another powerful tool in the identification of genes causing NTDs has been candidate gene studies. Although studies of candidate genes in pathways of folate metabolism and non–folate-related candidate genes also have not yielded any genes or genetic variants that are consistently associated with NTD risk, none of the genes that have been evaluated as NTD risk factors has been studied in sufficient detail to warrant its exclusion as a candidate. Lastly, epigenetic mechanisms may play a role in NTD causation. Epigenetic regulation of gene expression has been suggested to alter the gene function by several mechanisms; these include methylation of DNA at certain C-C dinucleotide repeat segments, with folic acid acting as a chief donor of methyl groups8 enzymatic modification of histone function9; and activation or suppression of gene function by transposons,10 also known as “jumping genes.” Since NTDs have been associated with complex patterns of inheritance and are influenced by environmental agents, it is logical to assume that epigenetic effects and not necessarily mutated gene sequences may be very important in the development of neural malformations. In addition to direct gene regulation, protein and amino acid structure and function can be directly altered, leading to malformations. Protein sequence, conformation, kinetics, modifications, and interactions can all be implicated in abnormal development. Profiles of several groups of functionally related proteins, including circulatory, cytoskeletal, and stress proteins and other proteins of unknown function, have been demonstrated to be important in the developmental regulation of neural tubes of mice and presumably NTDs.11 In the study of metabolic pathways, metabolomics, folic acid pathways have been implicated to influence the risk for NTDs.12 Studies of mutant enzymes in the folate metabolic pathway, particularly methylenetetrahydrofolate reductase (MTHFR), have suggested a possible association with NTDs.7–11 The likely role folate plays in the reduction of NTDs resides within the methylation cycle of homocysteine (▶ Fig. 2.2). Conversion of homocysteine to methionine occurs when donated methyl groups from folate utilize MTHFR and enzymes that require vitamin B12 as a cofactor. Homocysteine can act as a teratogen and may be important in human NTDs.13–19 The possible association with mutated MTHFR, cystathionine synthase, and altered homocysteine methylation further suggests the importance that folate metabolism may have in human NTDs.20 Fig. 2.2 Homocysteine methylation cycle. (Note: Homocysteine perturbs neurulation in experimental animals.) NTD, neural tube defect. Currently, whole-genome screening and the bioinformatic processing of genomic data hold the promise of improved molecular analysis of embryologic events. Whole-genome screening has been limited by cost (up to $10,000 per whole genome), poor understanding of the genome sequence, and the relevance of epigenetic effects., Reduced cost, high-speed genomic sequencing throughput, and better understanding of the genomic and epigenetic impact on disease will change the future of molecular embryology. During the morula and blastocyst stages, proper cellular orientation or polarity is initially established. By postovulatory day (POD) 5, the embryo has developed from a single fertilized egg, through morulation, and into a 32-cell blastocyst, with an eccentrically placed inner cell mass and a surrounding ring of cells called the trophoblast. The inner cell mass forms the epiblast and the hypoblast, similar to the dorsal (animal) and ventral (vegetal) poles in Xenopus.21 The epiblast ultimately forms the embryo. These first signs of cell polarity provide the fulcrum for the developing body axis and nervous system induction. The first obvious feature of rostrocaudal polarity occurs around POD 13, with thickening of the rostral end of the hypoblast into the prechordal plate and the birth of the primitive streak at the opposite end.21 The primitive streak elongates cranially in the midline of the embryo and becomes contiguous with the simultaneously developing primitive knot, or Hensen node, at the rostral end. The Hensen node functions as the embryonic “organizer” and is critical to induction of the notochord and neuraxis. A cascade of factors expressed in a spatially and temporally controlled manner tightly regulates the early steps in the development of the body axis. Numerous studies have uncovered several genes implicated in patterning of the primitive body axis; however, the Wnt1 β-catenin pathway is the most understood.22–24 Wnt1 activates a cascade that increases β-catenin, which directs early expression of target genes, such as brachyury and siamois (important for proper formation of the primitive streak25), cripto (responsible for proper dorsoventral polarity in the epiblast26), and twin (required for development of the embryonic organizer27). In addition, the Wnt1 β-catenin pathway influences bone morphogenetic proteins (BMPs),22–24 which play a significant role throughout neurogenesis but are especially critical during pregastrulation for helping to establish proper rostrocaudal polarity. BMPs subsequently serve as positive inducers of epidermal differentiation during primary neurulation. Other nonmolecular mechanisms also help to instruct proper axis determination. Proper perpendicular alignment of the embryo with the longitudinal axis of the uterine wall,28 pH and ionic gradients, cell orientation in response to the site of sperm entry into the egg, and/or gravitational forces22 may all affect future embryonic polarity. Clearly, mechanisms governing polarity during pregastrulation are not completely understood; several different processes are most likely involved. No known malformations occur during the pregastrulation period. Perturbations during these stages would affect the polarity and differentiation of early primordial stem cells, the ability of the embryo to implant properly, and the development of the placenta and critical extraembryonic membranes—all of which would be lethal to the early embryo. Establishment of the organizer, primitive streak, and proper embryonic axis marks the beginning of gastrulation, or formation of the three embryonic germ layers (▶ Fig. 2.3). During gastrulation, a dramatic reorganization of the embryonic cells occurs along the rostrocaudal, dorsoventral, and left–right axes. The initial relationship and orientation of cells established in the blastula are rearranged through coordinated cell movements. Around POD 16, successive waves of epiblast cells caudal to the Hensen node migrate into the primitive streak and form the prospective endoderm and mesoderm. Epiblast cells immediately around the Hensen node migrate into the node in the midline and then rostrally to form the notochord. Fig. 2.3 Gastrulation. (a) Schematic of embryonic axis determination. Key is the anteroposterior borderline, which determines the rostral–caudal border. (b) Cross section of a chick embryo demonstrating features of the three embryonic germ layers. AP, anteroposterior; BMP, bone morphogenetic protein; bFGF, basic fibroblast growth factor; L–R, left–right; RA, retinoic acid. The notochord, located between the ectoderm and endoderm, is vital for determining the rostrocaudal axis of the embryo beginning at the anteroposterior (AP) borderline, thought to occur in the region of the hindbrain–midbrain junction (see ▶ Fig. 2.3). The notochord also intercalates with the endoderm and contributes to the ventral floor plate region of the future neural tube. Another connection, previously known as the neurenteric canal, occurs in the pit of the Hensen node and connects the ectoderm to the endoderm.29 Several genes have been implicated in these early steps of gastrulation30; however, brachyury is clearly required for normal gastrulation movements and development of the three germ layers in embryos.31 As cells move away from the streak, brachyury expression is downregulated, except in the head region and notochord. Mice that are homozygously deficient in this gene do not form a primitive streak, have an absent or abnormal notochord, and lack caudal mesodermal structures.31 Downstream target genes of brachyury must include notochord-forming factors, cell adhesion molecules involved in gastrulation movements, and factors needed to continue expression of brachyury. Dorsoventral patterning is an important event during gastrulation and depends on the proper expression of brachyury as well as competitive interactions between ventral signals, such as sonic hedgehog (shh) and HNF-β,32 and dorsalizing factors from the ectoderm, such as BMP-4, Msx-1, and dorsalin-1.33–36 As discussed earlier, the formation of dorsal structures also requires the expression of Wnt/β-catenin factors.37 Establishment of left–right polarity is also thought to occur during gastrulation, with formation of the notochord; however, there is no anatomic correlate. Left–right asymmetry may be related to the asymmetric expression of growth and transcription factors during gastrulation. In avian embryos, activin-bB and its receptor are found only on the right side of the Hensen node, whereas shh is expressed only on the left. Similarly, in the mouse, nodal, a transforming growth factor-β (TGF-β) protein, and lefty are found only in the left-lateral mesodermal plate.30,38 Defects during gastrulation are thought to affect all three primary cell layers along all embryonic axes. Malformations include split-cord malformations (SCMs); neurenteric, dermoid, and epidermoid cysts; anterior and posterior spina bifida; intestinal malrotations, duplications, and fistulas; anterior meningoceles; and other complex dysraphic malformations. Additionally, congenital tumors, such as teratomas, may have their origin during gastrulation. Common to this class of defects is the presence of an SCM (▶ Fig. 2.4). SCM type I, formerly called diastematomyelia, comprises two dystrophic spinal cords separated by a vertical bony bar with two dural tubes. SCM type II, formerly called diplomyelia, comprises two dystrophic cords separated by a fibrous band contained in a single dural tube. Complex SCM, or type III, occurs when one of the dystrophic hemicords forms an open NTD, also called a hemimyelomeningocele. True dimelia, or complete duplication of a normal spinal cord. has not been described.39 These malformations represent a spectrum of defects of a common embryopathic mechanism. The open NTD associated with an SCM further underscores the likelihood that the SCM developed before the NTD. Fig. 2.4 Split-cord malformation. (a) Schematic showing the split notochord and the split ectoderm. (b) Myelogram demonstrating split cord malformation type 1. Many theories exist to explain the embryopathy of SCMs and more complex defects.40,41 The common theme is an underlying embryologic defect involving the formation of two notochordal structures. The notochord is split by a retained or accessory neurenteric tract, or it is split by the persistent separation of primitive paired notochordal anlagen. The separation of the notochord induces the formation of two separate neural plates, and retained tissue between the plates forms a tract or rest of pluripotential cells. Laterally displaced somites lead to vertebral anomalies such as butterfly vertebrae. The intervening tissue contains pluripotential cells capable of developing all three germ layers, including ectodermal malformations (tufts of hair, dermoid and epidermoid cysts); mesodermal malformations (bony, cartilaginous, or fibrous bands; muscle; adipose tissue; vessels); endodermal malformations (intestinal malrotations, neurenteric cysts); and lesions with all three germ layers, such as teratomas.42 The underlying defect remains unknown, but two basic mechanisms are likely: aberrant axial mesodermal cell movements and duplication of normal axial structures secondary to inappropriate instructive signaling. Aberrant cell movements have been described when the Hensen node is surgically split during late gastrulation in chick embryos.43 Inappropriate migration of the notochord has been reported in brachyury mutant embryos.31 Duplication of the caudal neural tube and notochord is caused by teratogenic levels of exogenous retinoic acid, antisense oligonucleotide inhibition of engrailed genes,44 or fibroblast growth factor receptor-1 (FGFR-1) manipulation.45 Primary neurulation is a complex morphogenetic process comprising several independent events that overlap temporally and spatially. The result is the development of the brain and spinal cord down to the S2 level. It can be divided into four major events: neural plate formation, neural plate midline bending, neural plate lateral wall bending, and neural fold fusion (▶ Fig. 2.5). Fig. 2.5 Primary neurulation. (a–c) Neural plate folding, elevation, and fusion. (d–f) Modes of neural tube closure. Mode 1 produces a V-shaped neural tube, mode 2 produces a diamond-shaped neural tube, and mode 3 produces an O-shaped neural tube. (g) Various initiation sites (sites 1 to 5) of neural tube closure. The primordial neural plate is first induced around POD 16 as a pseudostratified columnar epithelium.46 The process of induction occurs when a group of cells establishes, changes, or directs the development of an adjacent group of cells via direct contact or by the secretion of diffusible molecules.47 Neural induction is the default of ectoderm differentiation.36 Neuronal differentiation is actively inhibited in the ectoderm by ventral embryonic cells and early ectoderm.36 It appears that BMP-4 is responsible for neural inhibition and is blocked in the prospective neural ectoderm by antagonizing molecules, such as chordin, noggin, follistatin, and others.24,48–53 The newly induced neural plate is transformed from a flat, oval structure into a narrow, elongated one. Net cell movement is lateral to medial, along with intercalation in the midline to help lengthen the neural plate in the process known as convergent extension54 (▶ Fig. 2.6). Blocking the gene dishevelled can disrupt convergent extension.55 T-box genes similar to brachyury as well as Wnt pathway factors also appear important for proper convergent extension to occur.56,57 Cellular features accounting for transformation of the neural plate are increases in cell elongation and height, cell division, and cell rearrangement or intercalation. Fig. 2.6 Convergent extension, growth of the neural plate by medial then caudal cell movements. Around POD 17, a shallow midline neural groove in the neural plate forms directly above the notochord, initiating bending of the plate. Induced cells located in the midline of the neural plate change from a spindle shape to a wedge shape, forming a median hinge point (MHP).58 Cell wedging during neural plate bending is brought about by the contraction of circumferentially oriented apical microfilaments via a sliding mechanism utilizing actin and myosin,59 along with prolongation of the cell cycle.60 The notochord-secreted morphogen shh is the primary molecular substrate for this phenomenon.61 Mouse embryos exposed to cholesterol synthesis inhibitors are thought to produce holoprosencephaly and NTDs through an shh-mediated mechanism. Additional bending points, dorsolateral hinge points (DLHPs), occur in the cranial and low primitive spinal cord regions of the developing neural tube. The molecular determinants of DLHPs are largely unknown. DLHPs allow the tips of the neural folds to bend inward toward each other and converge.58 Neural fold fusion is poorly understood. Cellular mechanisms include interactions of cell surface glycoproteins, interdigitation of cell surface cilia, and formation of intercellular junctions. A surface coat of glycosaminoglycans (GAGs) becomes more concentrated at the fold tips immediately before fusion.62 The composition of GAGs changes during neurulation, with hyaluronic acid predominating before fusion and chondroitin sulfate predominating after fusion.62 Cell adhesion molecules (CAMs) and cadherin play a role in the separation of the neuroectoderm from the surface ectoderm. As neurulation proceeds, the neural plate stops expressing E-CAM and begins expressing N-CAM and cadherin. Cilia have become recognized as important in neural tube closure, likely as mediators of neural tube formation and neural fold fusion. It appears that cilia help mediate hedgehog signal transduction pathways.63 Gli-type transcription factor activity downstream is controlled by hedgehog and is important for the patterning of cell fate during embryogenesis.64 Animal models with mutant cilia developmental genes have disturbed neural tube closure.65,66 Recently, the MKS1 and MKS3 genes have been cloned in the Meckel-Gruber syndrome, which is known to have neural tube defects along with renal and limb abnormalities. The MKS1 and MKS3 genes are important in ciliogenesis, implying that cilia are important in neural development.67 Toward the completion of fusion, dorsal neural tube cells extend cellular protrusions medially and laterally, bringing about the radial intercalation of deep and superficial cells to form a single cell–layered, pseudostratified neural tube.68 Intercellular gaps or tight junction connections are established between cells on apposing neural folds, completing the fusion process.1 Human neural tube closure occurs between PODs 21 and 28, proceeding from three initiation sites along the neuraxis.69 Therefore, the fusion process occurs in multiple discontinuous waves until closure is complete. Genes such as PAX3 may be important in neural fold fusion. A paired-type homeobox gene, PAX3 is expressed in the lateral and posterior neural plate, and expression increases in the dorsal neural tube precisely at the time of neural fold fusion.70 Altered PAX3 function results in NTDs, as seen in the splotch mouse mutant.71 Extrinsic factors may also contribute to neural tube formation.72 These factors include compression of the neural folds by the medial convergence of the surface ectoderm, elevation of the neural folds by the accumulation of underlying mesodermal cells, expansion of the underlying extracellular matrix, and passive buckling of the neural tube by elongation of the notochord and floor plate of the neural tube.73 In addition, newly formed neural crest cells may contribute as they migrate toward the dorsal midline, with two lateral cell groups combining into a single medial population at the roof of the neural tube.68 Neural crest cell emigration itself may be a significant determiner of neural tube closure and subsequent dysjunction at the neurocutaneous interface. Neural crest cells are formed from a nidus of cells at the junction of the neural tube and surface ectoderm and migrate laterally after neural tube closure. The PAX3 and slug genes appear to be important in neural crest determination and migration. Human (Waardenburg syndrome)74 and mouse (splotch)75 mutants of PAX3 have defects in the migration of neural crest cells. When slug is inhibited in chick76 or frog,77 proper neural crest migration is blocked, and the neural tube fails to close. Neural tube formation is clearly more dynamic and complicated than initially proposed (see ▶ Fig. 2.5).78 At least three different modes of neural tube formation exist throughout the rostrocaudal axis, reflecting different mechanisms. Mode 1 occurs in the cervicothoracic region, where a distinct MHP forms without any evidence of DLHPs. The neural folds remain straight, resulting in an ovoid tube and slit-shaped canal. In the midbrain–hindbrain region, mode 2 occurs, with the appearance of a MHP and DLHPs. After fusion, the tube takes on a diamond-shape configuration, foreshadowing the shape of the fourth ventricle. Neural tube formation in the lumbosacral region, mode 3, involves only a suggestion of DLHP formation in addition to the well-developed MHP, forming a more circularly shaped tube with a large, patent canal. These neural tube differences may account for some regionally specific abnormalities. Before the neural tube completely closes, the cranial portion begins to undergo rapid expansion due to increased growth of the tube and enlargement of the ventricular system.52 Closure of the cranial neuropore with spinal canal occlusion isolates the ventricular system and increases the intraventricular pressure, which may drive brain enlargement.49 The anterior neuropore closes at about POD 24 as a thickening in the dorsal portion of the embryonic lamina terminalis, the future site of the anterior commissure.71 The caudal neuropore closes at about POD 26,49 corresponding to vertebral level S2. It appears that in humans, most of the spinal cord, as far as the S2 level, forms by primary neurulation; the filum terminale and lower sacral levels form by secondary neurulation. After neurulation is completed, the neural tube is clearly segmented along the AP axis into the various components of the future CNS. An important landmark in nervous system segmentation is the AP borderline that resides in the midbrain–hindbrain junction.79 Segmentation is not a function of neurulation but is determined earlier in development by the rostrocaudal gradients of homeotic gene expression. These gradients set in motion the sequential expression of sets of embryonic gap genes, pair rule genes, and segment polarity genes, each set resulting in progressively greater segmentation.80 Open human NTDs (▶ Fig. 2.7) include the common nonsyndromic anencephaly, cranioraschisis, myelomeningocele, and myeloschisis and reflect a spectrum of defects that occur during primary neurulation. Infants with anencephaly or cranioraschisis do not survive; therefore, these conditions are clinically irrelevant. However, it is imperative that these lesions be understood to discern how the clinically important myelomeningocele and myeloschisis develop. Four developmental mechanisms for open NTDs have been proposed based on data from humans and laboratory animal models: (1) factors important to intrinsic patterning to the neural plate, (2) factors extrinsic to the neural plate, (3) factors important along the rostrocaudal embryonic axis, and (4) reopening of a closed neural tube. Fig. 2.7 Open spinal neural tube defect. Note how the medial hinge region is normal and cutaneous ectoderm remains attached at the lateral borders of the neural plate. The first concept proposes that open NTDs are the result of an abnormality directly within the neuroepithelium. Studies of patterning and induction of the neural plate have revealed that ventral and dorsal neural plate structures are developed by specific but independent signaling cascades,81,82 and they are considered separately in the following paragraphs. The essential ventral cellular element for neural tube closure is the MHP. Ventral signaling molecules, such as shh83 and HNF-β,84 specify the formation of wedge-shaped cells required for neural plate folding. The phenotype of cranioraschisis in mutant animals, in which the neural tube is open from the level of the midbrain to the lower spinal levels, is thought to be the result of abnormal MHP determination. The telencephalon and midbrain close normally, indicating that the dorsal elements that lead to fusion are normal. In the four mouse mutants with cranioraschisis, dishevelled,85 loop-tail,86 circletail,87 and crash,88 the neural folds elevate normally but are widely spaced at the initial closure site. At the cellular level, abnormal neural plate development in cranioraschisis resulted from disturbed convergent extension, a consequence of disturbed Wnt signaling pathways.55 Disturbed convergent extension yields a shortened and broad neural plate, and thus a widened and misshapen MHP. The relevance of this model is that neurulation in the hindbrain and upper spinal cord depends on proper MHP development, and any abnormality affecting MHP development is likely to be lethal in humans. Critical dorsal neural plate determinates for neural tube closure are the proper formation of the DLHPs and the fate of roof plate precursors. Animal models with dorsal neural plate abnormalities have cranial defects, lower spinal defects, or a combination. The key mouse mutant for this model is the splotch-PAX3 mutant.75 In humans, PAX3 is defective in patients with Waardenburg syndrome, with a subset having spinal NTDs.74 The PAX3 gene is expressed in the presumptive dorsal neural plate, neural crest, and paraxial mesoderm that gives rise to skeletal muscles.89 In the splotch mouse, the MHP is normal, but the DLHP region is not. This has been confirmed in chick embryos treated with antisense oligonucleotides to PAX3.71 It is not known how mutant PAX3 causes NTDs; increased apoptosis,90 faulty pyrimidine synthesis, or alterations in cell migration91,92 have been proposed. In addition, altered function in neural crest precursors of the presumptive roof plate may alter neural tube closure. Splotch demonstrates repressed neural crest migration activity. Chick embryos treated with antisense oligonucleotides to PAX371 or slug,76,93 important in neural crest precursor migration, demonstrate open spinal NTDs. The second concept proposes that properties extrinsic to the neural tube are responsible for open NTDs. The best model is the curly tail mouse, which develops a lumbosacral myelomeningocele and is a phenocopy of nonsyndromic NTDs in humans.94 The inheritance, genetics, and response to periconceptual vitamins make this an attractive model for human caudal NTDs. At the tissue level, mutant curly tail embryos exhibit a cell type–specific abnormality of cell proliferation that affects the gut endoderm and notochord, and not the neuroepithelium.95 The reduced rate of ventral embryonic cell proliferation results in a growth imbalance between ventral gut primordia and dorsal neural elements. The result is a delay in posterior neuropore closure because of abnormal caudal flexion, resulting in spinal NTDs.96 The third concept is based on the location of the NTD as determined by rostrocaudal patterning. One common theme when the NTD pattern of the mutant models is analyzed is that the location of the defects appears to respect the restricted gene expression along AP axis domains established during gastrulation. The establishment of an AP borderline determines mesodermal and ectodermal tissue specification and gene expression. This transition zone exists between the midbrain–hindbrain boundary and is defined by rostral expression of otx genes and caudal expression by engrailed and hox genes.79 At the tissue level, the variation of gene expression along the rostrocaudal axis mirrors the variation in mechanisms of neural fold elevation, folding, and fusion (see ▶ Fig. 2.5). According to the model proposed by Shum and Copp,78 regional rostrocaudal differences in modes of neural tube closure will cause different types of open defects. Defective mode 1 causes cranioraschisis by interfering with MHP formation. Defects of mode 2 cause exencephaly due to defective DLHP function. Mode 3 defects cause open spinal NTDs. Additionally, many mutant mouse models have demonstrated that the number and location of the NTDs can vary along the neuraxis. For example, patch, PAX3, and short-tail mutants have defects localized caudal to the AP borderline, whereas apoB, hox-a1, and extra-toes mutants have defects rostral to the AP borderline. Several mutants with cranioraschisis have defects spanning the entire neuraxis. The last mechanism proposes that a properly neurulated neural tube can be reopened. The only spontaneous mutant in which this mechanism occurs is the curtailed mouse, in which increased cerebrospinal fluid pressure is thought to rupture a thinned roof plate and dermis in the absence of competent dorsal bony vertebrae.97 Although the curtailed mutant may indeed have a reopening of a previously closed neural tube, this mechanism seems unlikely in human NTDs. However, the reopening model is relevant for the neurosurgeon because the animal experiments that formed the basis for fetal repair of the myelomeningocele were predicated on a surgically generated “reopening” model.98 In the animal models tested, repair prevented intrauterine injury from either the toxic effects of amniotic fluid or direct intrauterine trauma.99 Regardless, no clear correlation can be made between the structural defect in these animal models and what occurs naturally in human myelomeningocele. Understanding the embryology of the myelomeningocele placode is of vital importance when novel repair strategies like intrauterine closure and spinal cord regeneration are considered. The placode has been described as a partially functional or nonfunctional remnant of the unneurulated spinal cord, and little is known about the cellular patterning or connectivity within the placode. The placode has preserved dorsal and ventral cord elements—for example, nerve roots and ganglia—but the exposed neural tissue demonstrates hemorrhages and abrasions indicative of injury.100 The placode exhibits abnormal cellular patterning along the dorsoventral and rostrocaudal axes, indicative of a change in pattern determination with a paucity of maturing neurons.101 Surgical repair will be predicated upon uncovering the true nature of the structure and function of the neural placode. Malformations like meningoceles, cervicothoracic meningoceles and myelomeningoceles, and meningocele manqué are completely or partially skin-covered and are not generally grouped with open defects.102 However, these lesions should be considered defects of primary neurulation. There is often evidence of limited dorsal myeloschisis in which the dorsal neural tube fails to fuse but neurocutaneous dysjunction occurs normally. Patients usually have normal neurologic function; therefore, it has been presumed that the patterning of the spinal cord is also normal. The molecular, cellular, and tissue-specific mechanisms for the development of these malformations are not known. Dermal sinuses and dermoid and epidermoid cysts can involve any level of the neuraxis, from the most rostral cranium to the caudal sacrum. Dermoid cysts include elements of mesoderm and ectoderm (skin, hair, sweat glands, and sebaceous glands), whereas epidermoid cysts are composed only of ectodermal elements (squamous epithelium). Dermal sinuses can be associated with tufts of hair. Whether or not to include coccygeal dimples in the spectrum of dermal sinuses is unclear. It is not clear if these are the remnant of the regressed primitive pit or a forme fruste of a dermal sinus overlying the caudal part of the spine. Since dermal sinuses can be associated with SCMs and completely neurulated CNS structures, it remains unclear if these are disorders of gastrulation or late primary neurulation. The most discussed mechanism involves incomplete dysjunction of the neurepithelium from the cutaneous dorsal ectoderm (▶ Fig. 2.8). It is thought that incomplete dysjunction occurs at one of the closure sites of the neural tube. There remains a persistent attachment between the neurepithelium and surface ectoderm. What governs dysjunction of the surface ectoderm from the neurectoderm after neural tube closure is not completely known, but several possibilities exist, as previously discussed. Fig. 2.8 Sinus tract formation; schematic of incomplete dysjunction of ectoderm after neural tube closure.
2.1 Introduction
2.2 Molecular Determinants of Congenital Defects
2.3 Embryogenesis of the Neuronal System and Malformations
2.3.1 Pregastrulation (Morula and Blastocyst Stages)
Defects of Pregastrulation
2.3.2 Gastrulation
Defects of Gastrulation
2.3.3 Primary Neurulation
Neural Plate Induction, Lengthening, Folding, and Fusion
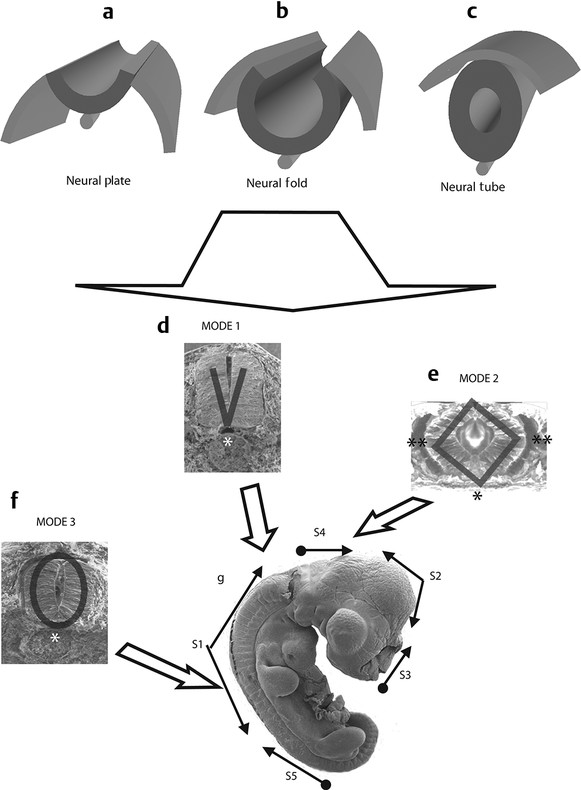
Variations in Neurulation along the Rostrocaudal Axis
Defects of the Primary Neural Tube (Anencephaly, Cranioraschisis, Myelomeningocele, Myeloschisis)
The Myelomeningocele Placode
Other Defects of the Primary Neural Tube (Meningoceles)
Defects Involving the Dorsal Ectoderm (Dermal Sinuses, Dermoid Cysts, Epidermoid Cysts)
Normal and Abnormal Development of the Nervous System
Only gold members can continue reading. Log In or Register to continue


Full access? Get Clinical Tree


