Brainstem Gliomas
Brainstem tumors exhibit substantial heterogeneity in their clinical attributes, imaging features, anatomical position, and histologic subtype and grade. Accordingly, the clinical behavior of these tumors can be equally variable but is relatively well defined based largely upon the anatomical growth pattern, a feature that parallels magnetic resonance (MR) imaging findings. It is therefore important to categorize a patient’s tumor based on this information in order to provide appropriate diagnostic and therapeutic recommendations.
42.1 Epidemiology
Brainstem tumors account for 10 to 20% of all central nervous system tumors in children.1–4 Of brainstem gliomas, approximately 80% are diffuse intrinsic pontine gliomas (DIPGs), which carry a very poor prognosis.1 However, roughly 15 to 20% are low-grade astrocytomas, and these follow a more indolent course. Approximately 150 to 400 children in the United States and 20 to 30 in the United Kingdom are thought to develop brainstem gliomas annually.1,3–7 The total incidence of brainstem tumors of all types rose from 0.31 per 100,000 between 1977 and 1981 to 0.67 per 100,000 between 1990 and 1994.5 This increase is primarily thought to be secondary to increased detection through the introduction of MR imaging rather than a true increase in the incidence of these tumors. There appears to be equal incidence regardless of gender. The most common age at diagnosis is 5 to 9 years, but cases have been seen in all age groups from infants to adults.6
42.2 Classification and Treatment Approaches
The classification of brainstem tumors is typically based on MR imaging.8 The overwhelming number of intrinsic brainstem tumors are astrocytic. Peculiarly, the histologic subtype and grade are fairly well predicted based on anatomical position or classification of the tumor. Tumors can be divided according to their location in the midbrain, pons, or medulla,8 and Epstein and Farmer suggested dividing tumors into three groups: cervicomedullary, focal medullary, and dorsal exophytic or diffuse anaplastic astrocytomas.9 Today, the classification of these tumors encompasses five principal and distinct subgroups: tectal (mesencephalic) tumors, DIPGs, focal tumors, dorsal exophytic tumors, and cervicomedullary tumors. Of course, not every brainstem tumor can be categorized as one of these defined entities, and the pathology may digress from the descriptive term. Because the current categorization scheme is defined by the anatomical features based on imaging, predicting the rare primitive neuroectodermal tumor (PNET) or ganglioglioma can be problematic, if not impossible. Therefore, any clinical or imaging aspects that raise suspicion about the conformity of the diagnosis should serve as an impetus for diagnostic sampling.
42.2.1 Tectal (Mesencephalic) Tumors
Approximately 5% of brainstem tumors are located in the tectal plate.10 They are most commonly indolent, slow-growing lesions limited to the rostral mesencephalon. Rarely, extension of the tumor into the posterior diencephalon occurs. They create symptoms primarily through the obstruction of cerebrospinal fluid (CSF) flow at the level of the cerebral aqueduct, resulting in symptomatic hydrocephalus.
On imaging, the tectal glioma is characterized by a thickened collicular plate with compression of the cerebral aqueduct. Resulting triventricular hydrocephalus is universally present unless the diagnosis is incidental. Because contrast enhancement is not likely, the mass is most easily seen on FLAIR (fluid-attenuated inversion recovery) and T2-weighted sequences on magnetic resonance (MR) imaging. The typical appearance includes hyperintensity on T2-weighted imaging and hypo- or isointensity on T1-weighted imaging (▶ Fig. 42.1).10 Lesions usually have poorly defined margins, although examples of focality have been reported.11 Exophytic growth patterns are infrequent, but infiltrative growth into one or both thalami can occur with some regularity. This lack of exophytic growth into the quadrigeminal cistern helps to distinguish most mesencephalic tumors from tumors originating from the pineal gland. However, for large midbrain tumors and those extending into the pineal region, serum alpha fetoprotein, β-human chorionic gonadotropin, and placental alkaline phosphatase should be measured in order to rule out primary CNS germ cell tumors (discussed in Chapter ▶ 39, “Pineal Region Tumors”).2 The pathology of midbrain tumors, if obtained, is typically low-grade astrocytoma or more rarely ganglioglioma.10,12
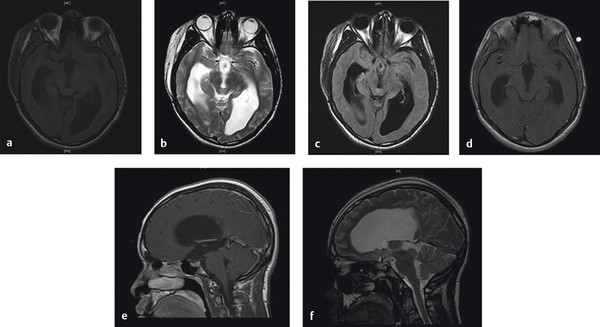
Fig. 42.1 Tectal mass. (a) Axial T1-weighted magnetic resonance (MR) image of a child with a tectal mass. Note significant resulting hydrocephalus. (b) Axial T2-weighted MR image and (c) axial FLAIR (fluid-attenuated inversion recovery) MR image for the same patient demonstrating hyperintensity. (d) Axial T1-weighted MR image with gadolinium contrast demonstrating typical lack of contrast enhancement. (e) Sagittal T1-weighted MR image with gadolinium contrast and (f) sagittal T2-weighted MR image.
Surgical Treatment
The surgical treatment of midbrain tumors is typically restricted to the treatment of hydrocephalus. Given the obstruction to CSF flow at the level of the cerebral aqueduct, the currently preferred method for CSF diversion in these patients is an endoscopic third ventriculostomy (ETV). Patients occasionally require a repeat ETV, but overall, the literature suggests that 80 to 100% of patients undergoing ETV in this scenario remain shunt-free with excellent control of the hydrocephalus.13–15 With respect to the tectal tumor, surveillance monitoring with serial MR imaging is generally used, with more frequent imaging (3 to 6 months) shortly after diagnosis and then annually if the mass remains stable.2,16 Biopsy of these tumors is reserved for patients who exhibit a rapid neurologic course, unusual imaging features, or progressive radiographic findings. It is not recommended that a routine simultaneous endoscopic tumor biopsy be performed at the time of ETV for patients with typical clinical and imaging findings. For those with tumors that ultimately progress, tissue sampling is recommended. Suitable surgical approaches are determined based upon the degree of ventricular enlargement, tumor growth pattern, and focality of the mass.
Nonoperative Management
Patients with midbrain tumors typically do well, and conservative management after the treatment of hydrocephalus is often the most appropriate course. However, approximately 18 to 31% of tumors will eventually require treatment.10,17 The size at presentation appears to have a significant correlation with the need for future treatment. A recent study showed that all tumors larger than 10 cm3 at presentation ultimately required treatment, whereas the majority of those smaller than 4 cm3 did not require intervention.10 However, some patients with tumors in the smallest size subgroup do ultimately require treatment (13.6% in this series), so surveillance is recommended even in these cases of seemingly benign lesions.10 For patients whose tumors progress, one of a variety of approaches may be employed, such as irradiation, chemotherapy, or subtotal resection with irradiation of residual tumor. Both irradiation and surgery appear to be extremely effective; Pollack et al reported regression or stabilization in patients treated with irradiation, and Ternier et al reported control with surgery and repeated surgery if needed.10,17 Ternier et al reported only one death in their cohort of 40 patients.10
42.2.2 Diffuse Intrinsic Pontine Glioma
DIPG arises from the pons, is infiltrative, and may extend into the midbrain or cerebellar peduncles.18 Patients who have DIPG typically present with focal neurologic deficits and develop hydrocephalus only as a later sequela. Cranial nerve abnormalities most commonly manifesting as ocular imbalance are commonly the first sign of these tumors. Other common cranial neuropathies include eye movement abnormalities, diplopia, dysphagia, dysarthria, and facial weakness or sensory loss.2 Head tilting to compensate for diplopia may be noticed in children who are too young to describe visual problems. Cerebellar dysfunction is another common presenting sign of these tumors, and limb weakness can result from involvement of corticospinal tracts.2,19
The MR imaging findings of DIPG are consistent with a hypocellular tumor that has more limited blood flow and angiogenesis than high-grade tumors. DIPGs are typically hypointense on T1 imaging and hyperintense on T2 and FLAIR imaging, with indistinct borders (▶ Fig. 42.2). Enhancement typically makes up only 0 to 25% of the tumor volume.18 Additional findings may include engulfment of the basilar artery or necrosis, but cysts are uncommon. MR spectroscopy shows a small decrease in N-acetylaspartate and an increase in choline, and MR perfusion demonstrates hypoperfusion.18 Atypical features include prominent enhancement, decreased T2 or FLAIR signal, restricted diffusion, and/or extensive exophytic components.
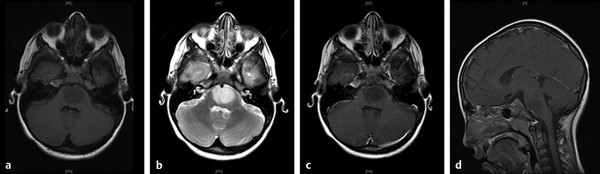
Fig. 42.2 Diffuse intrinsic pontine glioma (DIPG). (a) Axial T1-weighted magnetic resonance (MR) image of a child with DIPG demonstrating classic hypointensity and indistinct margin. (b) Axial T2-weighted MR image of the same patient demonstrating classic hyperintensity. (c) Axial T1-weighted MR image with gadolinium contrast demonstrating lack of enhancement. (d) Sagittal T1-weighted MR image with gadolinium contrast.
Although MR imaging is the standard modality for imaging brainstem lesions, advanced imaging modalities are under investigation and may prove useful in the future. A recent study of proton magnetic resonance spectroscopy in children with DIPG suggested that patients with an increased choline to N-acetylaspartate (Cho:NAA) ratio (thought to be suggestive of increased cell turnover, cell number, and neuronal damage) have a worse prognosis, and that an increase in this ratio from one time point to another is inversely associated with survival.6 This is consistent with studies in adult gliomas, which have shown that an increased Cho:NAA ratio correlates with a higher histologic grade.6 Positron emission tomography (PET) may also be of use in the assessment of DIPG, with two recent studies suggesting a trend toward a shorter length of survival in patients with increased fluorodeoxyglucose (FDG) uptake on PET.20,21 PET may also help in guiding stereotactic biopsy sampling because samples taken from areas with FDG uptake showed an equal or higher grade than areas without uptake, and in this small series the diagnostic yield was increased when PET information was used to guide the trajectory.20
MR imaging appears to be very accurate for classifying tumor type in cases with classic findings. In a recent study correlating imaging findings with histology in 44 patients with brainstem tumors, 100% of diffuse nonenhancing brainstem lesions (i.e., cases with imaging features typical of DIPG) were found to be consistent with astrocytoma with predominantly low-grade features (8 low-grade cases and 1 high-grade case). Of the enhancing diffuse lesions (atypical of DIPG), 90% were diffuse brainstem glioma, with a much higher proportion of high-grade samples in these biopsies (14 high-grade and 4 low-grade cases). The tumors of the two patients in whom other pathologies were identified included 1 ependymoma and 1 ganglioglioma.22
Surgical Treatment
Diffuse tumors are not resectable, and the role of biopsy in DIPG remains limited to tissue sampling for molecular therapeutic strategies or investigational purposes. Before the routine use of MR imaging, biopsies were performed to confirm diagnosis and provide information regarding prognosis. Since the advent of MR imaging, DIPG can often be diagnosed without biopsy. Albright et al argued that biopsies were frequently not indicated in children with brainstem lesions because the lesions could be diagnosed and treated solely on the basis of MR imaging without subjecting the children to the risk of biopsy.23 An important aspect of diagnosis today involves differentiating between “classic” DIPG and “atypical” DIPG; “classic” DIPG is typically diagnosed solely on imaging, whereas biopsy may play a role in “atypical” DIPG in order to rule out alternate diagnoses. Additionally, as more molecularly specific treatments are developed, the likelihood that biopsy may alter treatment and the need for biopsies for this reason should increase. The information gained through the biopsy and characterization of these tumors could prove critical to a better understanding and treatment of these neoplasms. Although many neurosurgeons would not biopsy a “typical” DIPG, considerable variability exists between pediatric neurosurgeons as to whether an individual case is typical or atypical and whether or not they would consider biopsy of a lesion.24 A range of biopsy approaches are used depending on tumor characteristics and surgeon preference, including stereotactic suboccipital–transcerebellar biopsy, stereotactic supratentorial–prefrontal biopsy, and open biopsy.24,25 Most historical series investigating risk of biopsy were done with computed tomography (CT)–guided imaging. They suggest a 0 to 4% mortality rate, a 0 to 4% permanent morbidity rate, and a 0 to 28% transient morbidity rate.26–32 The most recent study suggests 0% mortality, 0% permanent morbidity, and 8% transient morbidity.31 Today, MR imaging–guided biopsy has replaced CT-guided biopsy. With MR imaging–guided stereotactic biopsy, the risk for mortality and morbidity was recently estimated to be between 0 and 6%.18 Of note, parents of children with DIPG seem to be willing to allow biopsy, with a recent study showing that 24% of parents would consent to biopsy for the sole benefit of future treatment development without any direct benefit to their child and that 55% would consider biopsy if there were only a slight chance of benefit.18 Thus, given the low morbidity of biopsy with current stereotactically guided techniques, biopsy may increasingly be indicated as part of clinical trials and as an aid to treating patients with specific molecular therapeutics. Additionally, biopsy may be indicated in diffuse lesions with atypical features in order to rule out other types of brainstem lesions that may be more amenable to resection or other therapy.
Radiation and Chemotherapy
Standard conventional radiation therapy consisting of daily fractions given 5 days a week for up to 6 weeks is the standard of care for DIPG, with a total dose of approximately 54 Gy.1,2,16,18 Most literature suggests the prolongation of progression-free survival but not overall survival with radiation, although a recent study suggested a small survival benefit in patients treated with radiation.7,33,34 Hyperfractionated and hypofractionated regimens have not shown any significant benefit in overall survival when compared with standard conventional radiotherapy.1,7,18 The risk for brainstem injury appears to increase at doses greater than 64 Gy.18 Overall, radiation has an 85% clinical response rate but only a 50% radiologic response rate.1 The clinical and radiologic responses do not directly correlate, and the relevance of the radiologic response is not clear.1 Additionally, for most patients, radiation provides only a transient improvement or stabilization of symptoms, with progression of disease about 8 to 9 months after diagnosis.16,18 Once tumors progress, there is a tendency for rapid deterioration, with the median time between progression and death ranging from 1 to 4.5 months.1 A recent study also suggested that radiation had similar effectiveness when delayed by upfront chemotherapy.35
Most studies of chemotherapy for brainstem lesions have focused on DIPG, yet chemotherapy has not been shown to offer a clear survival benefit when used alone or in conjunction with radiotherapy in these tumors.1 Assessment of chemotherapy results has also been limited by varying patient populations and criteria for response in the different trials, making a comparison of results difficult.1 One study suggested a possible benefit of chemotherapy in patients in the HIT-GBM database (primarily a German database). The study found that patients treated with chemotherapy showed an increased median survival compared with those treated without chemotherapy, yet most patients who did not receive chemotherapy also did not receive radiation. When only patients treated with irradiation were included, patients treated with chemotherapy and radiation did have a longer survival than those treated with radiation alone.34 However, this study included patients treated with several different chemotherapy regimens, and it is not clear whether the populations of patients who received chemotherapy and who did not are comparable. Another study, which demonstrated the longest published median survival of 17 months, suggested that preradiation chemotherapy may extend survival.35 In this study, BCNU (bischloroethyl–nitrosourea), cisplatin, and in some patients tamoxifen were alternated with two courses of high-dose methotrexate before radiation. Patients then received standard radiation with concurrent hydroxyurea. However, the cost of chemotherapy was high, with significantly longer hospital stays and multiple serious infections.35 Additionally, the patient cohort had a mean duration of symptoms of approximately 2 months before the initiatoin of therapy, and a longer prodrome has been associated with better survival. Further clinical trials with varying chemotherapy regimens are currently under way.
Future directions include chemotherapeutic agents or therapy with specific molecular targets and the stratification of chemotherapy based on the presence of these targets in a patient’s tumor. Two recent Phase I studies of imatinib used this strategy. Imatinib (Gleevec/Glivec; Novartis, East Hanover, NJ) inhibits PDGFR, ABL, and c-KIT tyrosine kinases by inhibiting the adenosine triphosphate (ATP)–binding site. These studies looked at KIT, PDGFRA, and PDGFRB expression in the tumors of patients who were then treated with imatinib.36,37 Although neither study demonstrated a clear response of tumor to drug, both had individual patients with stable disease, and they provide an important blueprint for testing chemotherapeutic agents in conjunction with assessing the presence of their molecular target. A previous Phase I trial of imatinib without molecular stratification also had two patients who appeared to respond, but the majority of patients did not exhibit a response.38 The molecular status of these two patients is unknown. Of note, imatinib has poor penetration of the blood–brain barrier; the second-generation tyrosine kinase inhibitor dasatinib (Sprycel; Bristol-Myers Squibb, New York, NY) shows much better permeability.37 Another molecularly targeted therapy, gefitinib (ZD1839, Iressa; AstraZeneca, Wilmington, DE), an EGFR tyrosine kinase inhibitor, was recently tested in a Phase II study, and patients treated with this agent appeared to show slightly more positive survival and progression-free survival rates than those of historical controls.39 Given the variability of brainstem tumors, it is impossible to determine whether the children in this study are similarly representative of those in historical studies. Thus, the effect could be either over- or underestimated. However, these patients did not undergo biopsy, so EGFR expression in the tumors treated is not known, raising the possibility that the results might improve significantly if only patients with EGFR expression were treated. EGFR can also be targeted via monoclonal antibody, and nimotuzumab, a humanized anti-EGFR monoclonal antibody, appears promising in preliminary clinical studies.40 Future trials should focus both on molecular stratification and efficient delivery of chemotherapeutic agents in order to achieve the best results. Additionally, combinations of molecularly targeted chemotherapies may be more beneficial than treatment with individual agents.
Therapeutic Advances (Molecular Characterization, Vaccine Therapy, Convection-Enhanced Delivery)
Understanding the biology of DIPG and other brainstem tumors is an important step in developing more effective treatments. Most research on brainstem tumors has been focused on DIPG, given its grim prognosis; however, the study of DIPG has been limited by a lack of tissue due to the difficulty of operating in this area. Brainstem biopsies were first suggested in the late 1970s and 1980s, but by the 1990s, biopsies were discouraged in typical cases of the disease. Consequently, tissue samples have primarily been restricted to autopsy samples. However, recently some progress has been made in defining the molecular characteristics of DIPG. This is critical for deriving appropriate chemotherapeutic strategies. Previously tried drugs have primarily been those used for adult glioblastoma, but DIPG appears to have distinct biological differences from adult glioblastoma multiforme and even from pediatric supratentorial high-grade glioma,41–43 with the exception of some cases of pediatric midline/thalamic supratentorial high-grade glioma, which may have some similarities.43
Zarghooni et al undertook whole-genome profiling of 11 patient samples of DIPG. Their results may not be fully representative because these were primarily autopsy specimens. Nonetheless, some important molecular characteristics of DIPG began to emerge. In their study, 50% of DIPG samples showed gain of PDGFA or PDGFRA. Additionally, all samples showed expression of PDGFR-α and downstream target phospho-mTOR (mammalian target of rapamycin).41 Becher et al similarly found a high rate of expression of PDGFR-α in high-grade brainstem glioma samples, with 87.5% of surgical samples and 50% of autopsy samples showing immunopositivity (overall rate, 67%).44 Paugh et al found gains of receptor tyrosine kinases in 56% of their DIPG samples and reported that gains of PDGFRA were seen in 30%. They found overexpression of PDGFRA in tumors with amplification and even in a subset of tumors without amplification.42 These results suggest that the PDGF pathway may play an important role in DIPG. This is further supported by the induction of brainstem gliomas in mice through overexpression of PDGF in the posterior fossa, and the establishment of a PDGF-induced brainstem glioma mouse model that recapitulates many of the features of brainstem glioma.44 Other receptor tyrosine kinases seen to have recurrent gains in DIPG (although with a lower frequency than PDGFRA) are MET, IGF1R, ERBB4, and EGFR.42 Of note, overexpression of IGF1R, PDGFRA, and EGFR has also been seen in cases without amplification of the genes.41,42 The role of the EGFR pathway is less clear, with one study suggesting that it is less frequently involved in DIPG than in adult supratentorial glioblastoma multiforme, and another study suggesting that ERBB1 is amplified and overexpressed in high-grade DIPG samples.41,45 Additionally, another recent series with a small number of samples found that 40% of samples demonstrated expression of epidermal growth factor receptor variant III (EGFRvIII), the most common variant of EGFR, which is expressed in many tumor samples but rarely in normal tissue.46 This finding has been used as the basis for a Phase I trial of an EGFRvIII peptide vaccine (a vaccine previously trialed in adult patients with glioblastoma multiforme) after conventional radiotherapy in patients with DIPG.46
Genomic analysis suggests that the transcription factors and genes associated with developmental processes are most significantly differentially expressed in DIPG versus cerebral pediatric high-grade glioma. In particular, members of the HOX family, critical genes for correct development, including HOXA3, HOXA2, HOXD3, HOXB2, and HOXD4, show significantly higher expression in DIPG.42 Loss of heterozygosity analysis has suggested that mutations in DNA repair pathways may also play an important role in DIPG, and poly (ADP-ribose) polymerase-1 (PARP1), a DNA repair gene that is critical for tumor survival in patients with compromised DNA repair ability, was gained in several tumor samples.41 PARP protein was seen to be expressed in additional samples, and PARP inhibition could be a potential therapeutic target.41
One study looking at genetic alterations found that loss of 11p, 17p, 14q, 18p, and 22q was more frequent in DIPG than in pediatric supratentorial high-grade gliomas. Gains of 17q and 10p were unique to DIPG, whereas gains of 1q and 9q were common to DIPG and pediatric high-grade supratentorial gliomas in other locations.41 Another study with a larger number of samples found DIPG had more frequent gains of chromosomes 2q, 8q, and 9q and losses of 16q, 17p, and 20p than non-brainstem glioblastoma. Chromosome 7 gain and 10q loss were comparable in DIPG and non-brainstem pediatric glioblastoma but less frequent than in adult glioblastoma multiforme, and gain of 1q was similar in DIPG and pediatric glioblastoma multiforme but more frequent in both than in adult glioblastoma multiforme. Loss of 13q and 14q was similar in glioblastoma multiforme in all ages and locations.42 A third study showed that loss of 17p and 14q was more common in DIPG than in pediatric high-grade glioma, and gain of 1q was seen at approximately similar rates in both DIPG and pediatric high-grade glioma.47 These studies share the findings that loss of 17p is more common in DIPG than in other gliomas and that gain of 1q is common to both pediatric DIPG and supratentorial glioblastoma multiforme. Of note, 17p is the location of the TP53 tumor suppressor gene.
Puget et al recently published their results analyzing DNA and RNA from 32 and 23 patients, respectively, with biopsies taken at time of initial diagnosis, the largest sample size to date.43 They suggest that DIPG can be subdivided into two groups that, although different from adult glioblastoma multiforme, have similarities to the proneural and mesenchymal variants of glioblastoma multiforme, respectively. The first group is characterized by overexpression of oligodendroglial markers and strong expression of Olig2 by immunohistochemistry. The gene expression profile of this group was significantly enriched for PDGFRA and the genes associated with this receptor. Additional receptor tyrosine kinases were sometimes also amplified. The survival of this group was significantly worse than that of the second group; 70% of children died before the median overall survival of 10.6 months, whereas 10% of the second group died before this median survival. When samples were compared based on histology alone (allowing the inclusion of 55 samples), patients with oligodendroglial features had a 7.73-month overall survival, compared with a 12.37-month survival for those who had primarily astrocytic features. DIPG in the patients in group 2 was characterized by the upregulation of mesenchymal transition genes, including master epithelial–mesenchymal transition regulators SNAI1 and SNAI2/Slug genes, overexpression of proangiogenic genes, endothelial proliferation, and activation of the HIF1A pathway. Group 2 tumors had a higher expression of STAT3, a gene whose elimination promotes neurogenesis and inhibits astrogenesis, and these tumors also appeared to be more similar to fetal neural stem cells.43 Additionally, numerous homeobox genes and genes in the sonic hedgehog pathway were found to be overexpressed in brainstem gliomas as opposed to supratentorial high-grade gliomas, and LHX2 and IRX2, two genes that have been described to be preferentially overexpressed in posterior fossa pilocytic astrocytoma and ependymoma compared with supratentorial tumors of the same histology, were also seen to be overexpressed in DIPG compared with supratentorial high-grade glioma. This suggests that there may be gene expression profiles in glial tumors related to the location of origin regardless of histologic diagnosis,43 an idea first proposed for ependymoma and pilocytic astrocytoma. Recently, Monje et al suggested that a nestin+/vimentin+/Olig2+ cell population in the ventral pons may be the cell of origin of DIPG and that a similar cell population in mice was increased upon upregulation of the sonic hedgehog pathway.48
The most recent avenue of genetic research involves a focus on histone H3 mutations. Histone lysine methylation is thought to play an important role in regulating gene expression and chromatin function, and H3.3 is the major histone associated with chromatin during brain development. Two recent studies demonstrated that more than 70% of DIPGs had a lysine-to-methionine mutation on histone H3.3 (K27M-H3.3) or the related H3.1.49,50 Additionally, it was found that patients with the K27M-H3.3 mutation have a significantly worse prognosis than patients with wild-type H3.3, and that PDGFRA gain or amplification is seen only in patients with this mutation, suggesting that histone mutation is an early event in the development of DIPG.50
An additional avenue of research is that of improving therapeutic delivery. The effectiveness of the systemic delivery of chemotherapy for brain tumors is significantly inhibited by the blood–brain barrier. Even a drug that is effective in the laboratory against glioma may not achieve high enough concentrations in the brainstem to have an effect. Thus, research addressing improved delivery to the brainstem is critical, in addition to research investigating improved chemotherapeutic agents. One such delivery method is interstitial infusion, or convection-enhanced delivery (CED). In this method, an agent, such as an antibody- or tumor-targeted toxin, may be infused through a small cannula directly inserted into a tumor in order to deliver the agent throughout the tumor. Brainstem tumors, with their focal location and poor prognosis, seem an ideal target for this therapy.51 Studies in rats and primates have suggested that agents can safely be delivered to the brainstem via CED.52–54 Additionally, a radiation-induced recurrent glioblastoma in the brainstem of a patient in Japan was found to regress after CED of nimustine hydrochloride.55 Currently, clinical trials are under way to determine the safety and efficacy of delivering specific agents via CED in pediatric patients with DIPG.
Overall, a new understanding of the biology of brainstem tumors will help in the development of improved therapeutics, and stratification based on tumor biology may help identify subsets of patients who might benefit from currently available therapies. The combination of research into new therapeutics and new delivery methods offers the best hope of an improved treatment strategy for these currently challenging tumors.
42.2.3 Focal Brainstem Tumors
Focal brainstem tumors may be located anywhere within the brainstem but are not usually located within the ventral pons. Focal tumors have well-defined margins typically best appreciated on T2 or FLAIR imaging and occupy less than 50% of the axial diameter of the brainstem.22 They are typically hypointense on T1 imaging and hyperintense on T2 imaging, and they have variable contrast uptake. They may be solid or cystic; however, the area of tumor identified on T1 imaging is usually equivalent to the area identified on T2 imaging.56 Focal tumors may present with headache, diplopia, swallowing difficulties, long tract signs, or other cerebellar or cranial nerve dysfunction depending on their location. Symptom duration is typically longer for low-grade tumors and shorter for higher-grade tumors. Focal brainstem gliomas are typically pilocytic astrocytomas or other low-grade gliomas.22,57 Less frequently, they may be gangliogliomas or high-grade gliomas.22,57
Surgical Treatment
An attempt at surgical resection should be considered for tumors that are clearly focal on imaging. The surgical approach must be tailored to the location of tumor. A study of 34 patients with focal brainstem lesions undergoing surgery demonstrated 0% postoperative mortality, 15% morbidity, and symptom improvement in 65% of patients.56 The selection of patients with clearly focal tumors and surgeon experience are both key to minimizing surgical morbidity. Neurophysiologic monitoring is also helpful in facilitating resection. For patients with tumors that are not clearly characterized as focal or diffuse through MR imaging, one strategy is to perform stereotactic biopsy and subsequently consider resection if the pathology demonstrates pilocytic astrocytoma, other low-grade astrocytoma, or ganglioglioma.
Radiation and Chemotherapy
Radiation or chemotherapy may be indicated for patients with unresectable lesions or for those with residual tumor after subtotal resection. Gamma knife radiosurgery has also been used to treat focal lesions; one study of 20 patients demonstrated an 84% progression-free survival rate at 5 years.58 However, the mean duration until tumor shrinkage was 14.8 months. Tumor response was correlated with higher Karnofsky performance score, higher peripheral dose, smaller tumor volume, and longer symptom duration before radiation on univariate analysis and with Karnofsky performance score alone on multivariate analysis.58
42.2.4 Dorsal Exophytic Tumors
Dorsal exophytic tumors are frequently found within the medulla, although they may also arise from the pons and pontomedullary junction. They protrude from the brainstem dorsally into the fourth ventricle. On MR imaging, they typically are hypo- or isointense on T1 and hyperintense on T2, and they show contrast enhancement (▶ Fig. 42.3).12 Patients frequently present with signs and symptoms attributable to hydrocephalus, such as failure to thrive or headache and vomiting, and are more likely to have a longer symptom duration before diagnosis than patients with other types of brainstem tumors.12 Papilledema, nystagmus, and cranial nerve dysfunction are also common findings. Long tract signs are not typically seen. Histology typically demonstrates low-grade (World Health Organization [WHO] grade I or II) astrocytomas or gangliogliomas.2,12
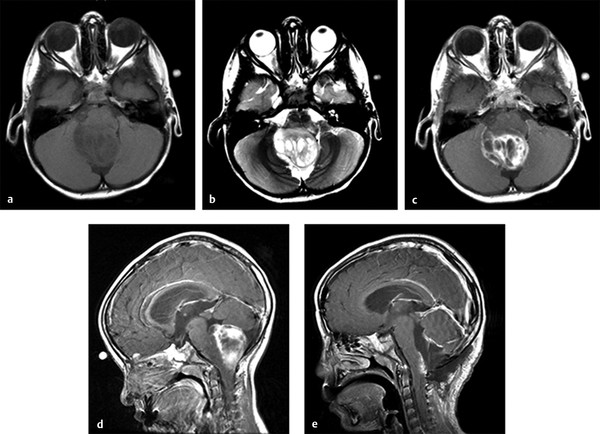
Fig. 42.3 Dorsal exophytic brainstem tumor. (a) Axial T1-weighted magnetic resonance (MR) image of a child with a dorsal exophytic brainstem tumor demonstrating dorsal extension of the tumor. (b) Axial T2-weighted MR image of the same patient. (c) Axial and (d) sagittal T1-weighted MR images with gadolinium contrast demonstrating contrast enhancement, as is typical of this type of tumor. (e) Sagittal T1-weighted MR image with gadolinium contrast of the same patient after resection. Most of the exophytic tumor could be resected. However, these tumors typically have a poorly demarcated interface with the brainstem, so that a small amount of tumor must be left to prevent injury to the brainstem. Note the area of contrast enhancement representing residual tumor on the dorsal surface of the brainstem.









