Cerebellar Astrocytomas
Except in infants and very young children (younger than 1 year), the posterior fossa is the most common site of occurrence for pediatric primary brain tumors.1 Of the tumors occurring in this location, medulloblastoma is the most common, accounting for 35 to 50% of cases and presenting mainly in the first decade of life,2,3 followed by cerebellar astrocytomas, which can develop throughout childhood and into early adulthood.4,5 Historically associated with a favorable outcome, individual cerebellar astrocytomas in reality can display different behaviors, depending mainly on the presence or absence of invasion, extent of surgical resection, and histologic grade. An understanding of the natural history of the disease, as well as the available therapeutic options, is necessary to achieve the best outcome in each case.
43.1 Epidemiology
Cerebellar astrocytomas occur most often in the pediatric population, with 70 to 80% of cases developing in children.6,7 They account for 10 to 20% of all pediatric brain tumors2,7 and up to 25 to 35% of posterior fossa tumors in children,6,8 with an incidence of 0.3 to 0.4 cases per 100,000 children per year.8 The mean age at diagnosis is 6 to 7 years7,9,10 when patients younger than 18 years are considered, and it is 17 to 18 years when adult cases are taken into account.7 Only 3% of cases have been reported in children younger than 1 year.11 Whereas medulloblastoma more frequently affects males, cerebellar astrocytomas show no evidence of a gender predilection.5,7,9,10 Historically, pilocytic astrocytoma has been reported as the most frequently occurring tumor, accounting for 70 to 80% of cases, followed by low-grade diffuse astrocytoma (10 to 15%) and higher-grade tumors, such as grade III anaplastic astrocytoma and grade IV glioblastoma multiforme (5%).6,12,13 However, if the recently described new entities, such as pilomyxoid astrocytoma and “diffuse” pilocytic astrocytoma (see below), are taken into account, up to 95% of all cerebellar astrocytomas consist of pilocytic and pilomyxoid variants, whereas the remaining 5% consist of diffuse astrocytomas of both low and high grades.14 The cerebellum is the most common location for pilocytic astrocytoma in children,15 except in patients affected by neurofibromatosis type 1 (NF-1), in whom cerebellar involvement occurs in fewer than 1% of cases.16
43.2 Pathology
43.2.1 Gross Appearance
Pilocytic Astrocytomas
Pilocytic astrocytomas (PAs) are very common World Health Organization (WHO) grade I tumors. They grow slowly and have a relatively low propensity for parenchymal infiltration. Often, cerebellar PAs invade the subarachnoid space, but this is not associated with an increased risk for cerebrospinal fluid (CSF) spread or a worse prognosis.17 Distant leptomenigeal dissemination has been occasionally described and may be (although not always) associated with a worse prognosis, as long-term survival occurs even without ancillary therapy.14,18–21 Thus, CSF dissemination may indicate, for example, simple chance fragmentation and dispersal of tumor already present in the subarachnoid space. PAs usually appear gray to tan–pink and may have pronounced vascularity.17 Tumor consistency varies from very soft and gelatinous to firm and rubbery (rarely, rock-hard). Lesions in the cerebellar hemispheres often show one or more macrocysts10,11 filled with proteinaceous fluid and delimited by a cyst wall that can be either neoplastic or formed by reactive gliosis.7,10,22 Macrocysts may occur adjacent to tumor or within tumor. The tumor often appears as a solid mass (“mural nodule”) in the cyst wall. Tumors of the vermis or fourth ventricle are usually less macrocystic.10 Some PAs appearing to “arise within” the fourth ventricle are actually dorsal exophytic growths from the brainstem that enter the fourth ventricle through the outflow foramina. Calcification occurs occasionally. Hemorrhage also occurs occasionally, and evidence of prior hemorrhage is the presence of perivascular hemosiderin pigment. In a rare case, acute hemorrhage may occur into a macrocyst, causing sudden decompensation with increasing mass effect and intracranial pressure (ICP), edema, herniation, and death.
Pilomyxoid Astrocytomas
Pilomyxoid astrocytomas (PMAs) were characterized in 1999.23 This tumor was included in the 2007 WHO classification as an independent grade II entity, particularly because of its tendency to more aggressive behavior compared with PA.14 PMAs are most often seen in the first 2 to 3 years of life and occur mostly in the midline, especially in the region of the optic chiasm, hypothalamus, and third ventricle; in this midline supratentorial region, PMAs may occur in older children, but cerebellar PMAs are generally limited to those younger than 2 to 3 years old. Currently, PMA is believed to represent a more aggressive “variant” of PA, with a greater tendency to local recurrence and CSF dissemination versus PA. This appears especially true of PMAs that arise in the supratentorial midline.14,23 Progression-free survival (PFS) is shorter for PMA than for PA, and “death from disease” is more frequent with PMA. Grossly, PMAs are well circumscribed, solid (a small fraction may have a usually minor cystic component), pale tan–pink, soft, and gelatinous. They may also be partly very soft, pale yellow, and necrotic (a small fraction, but more of them than of PAs).
Diffuse Fibrillary Astrocytomas
Diffuse fibrillary astrocytomas (DFAs) are relatively infrequent WHO grade II, grade III (anaplastic astrocytoma), or grade IV (glioblastoma, glioblastoma multiforme) tumors in the cerebellum. They appear as gray–tan–red, ill-defined, infiltrative lesions, with a consistency ranging from firm to gelatinous. These tumors may display microcystic degeneration, but they usually lack the frank macrocystic appearance of many PAs. Glioblastoma multiforme (GBM) characteristically has central necrosis and may show gross thrombosis of associated larger vessels.
43.2.2 Microscopic Appearance
Pilocytic Astrocytomas
The most classic feature of PAs (▶ Fig. 43.1) is the “biphasic pattern.” This is characterized by dense pink–red fibrillary regions composed of many spindle-shaped bipolar (piloid) astrocytes with elongated nuclei and very long and thin hairlike processes extending from both tips of tumor cells. These regions alternate with intervening looser areas that represent microcystic and often mucinous degeneration of the tumor (with cellular elements composed predominantly of multipolar rather than bipolar cells, cells resembling “protoplasmic” astrocytes, or even oligodendroglioma-like cells with generally more round to oval nuclei).
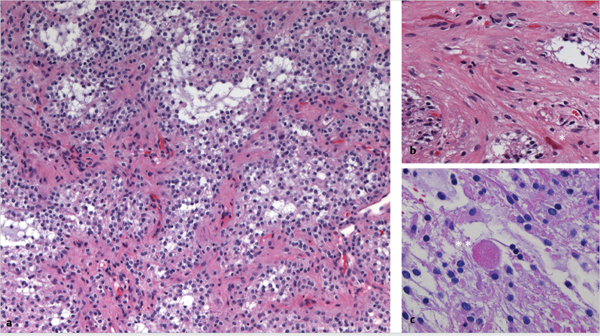
Fig. 43.1 Histologic appearance of pilocytic astrocytoma. (a) Low-power microscopic field after hematoxylin and eosin staining shows the classic biphasic pattern of dense eosinophilic fibrillary areas (pink–red) intermixed with loose mucinous microcystic regions (pale gray–white). (b) At higher magnification, Rosenthal fibers (single asterisks) appear as thick, dense, glassy, and eosinophilic corkscrew-like structures, found mainly in the dense fibrillary areas, whereas (c) eosinophilic granular bodies (double asterisks) appear as round pink granular structures, mainly localized to microcystic areas.
Other forms of degenerative change seen in PA include increased numbers of hyalinized (thickened and sclerotic, or scarred) blood vessels, occasionally associated with downstream foci of bland ischemic-type tumor necrosis, possibly related to occlusive events occurring within these abnormal vessels, and often marked yet benign nuclear atypia or pleomorphism; both hyalinized vessels and “degenerative”-type nuclear atypia are common in PA. The latter may appear worrisome (resembling anaplasia to the inexperienced), but these markedly atypical nuclei express no proliferation markers, show no mitotic activity, and occur in a background of otherwise typical features of PA. In addition to increased nuclear atypia, foci of necrosis, and extension into the subarachnoid space, other features that may appear worrisome to the less experienced eye include rare scattered mitotic figures, glomeruloid-type vascular proliferation (usually capillary-size vessels with multiple lumina and often only simple hypertrophic endothelium, but occasionally intraluminal hyperplasia or multilayering of endothelial-type cells, the latter tending to occur adjacent to macrocysts), and sometimes relatively increased cellularity. None of these aforementioned features have any prognostic significance in PA.14,24 Subarachnoid spread of PA is evidenced by a micro-multinodular pattern of growth, with variably thin fibrous septae present between small tumor nodules.
Other often helpful diagnostic features, which alone are neither necessary nor sufficient for the diagnosis of PA, include Rosenthal fibers (dense, glassy, hyaline pink–red intracytoplasmic inclusions, seen as corkscrew-shape structures in longitudinal sections or as discrete round to ovoid bodies in cross sections and mainly concentrated in the denser pink fibrillary areas of the tumor) and eosinophilic granular bodies, also intracytoplasmic and round but granular pink–red, raspberry-like protein aggregates, typically better seen in looser microcystic/mucinous foci.25,26
Rosenthal fibers are also seen in reactive (“piloid”) gliosis and thus are not always a sign of neoplasia. In piloid gliosis, the number of Rosenthal fibers present usually is greater than the number of nuclei seen, and microcystic change is also not generally present.
Pilomyxoid Astrocytomas.
The microscopic appearance of PMAs (▶ Fig. 43.2) is characterized by the following: a markedly predominant to solely (i.e., monophasic) “loose” microcystic/mucinous or myxoid background pattern, without any significant dense fibrillary component; a frequent, characteristic, often prominent vasocentric pattern of tumor cells attaching to and surrounding blood vessel walls; typically uniform, monomorphic (piloid) bipolar spindle tumor cells; and essentially complete absence of Rosenthal fibers, as well as a severe dearth or absence of eosinophilic granular bodies.23,25 Tumor cells are characteristically arranged in a perpendicular fashion around blood vessel walls, revealing a so-called vasocentric or perivascular rosette-like configuration.27 These perivascular collars are usually composed of a single layer of elongated tumor cells, which insert relatively stout processes into the vessel walls. Necrosis is not common but occurs somewhat more frequently than in PA. Calcification is relatively rare in PMA and is occasionally seen in PA. The mitotic index in PMA may be slightly higher than that in PA.25 Occasionally, glomeruloid-type microvascular proliferation may be seen in PMA. Some PMAs appear to “mature” (or perhaps “burn out”) into PAs over time,28 but initial sampling error (i.e., secondary to the receipt of only a small biopsy specimen for pathologic evaluation) may also explain this “phenomenon,” in at least some cases. Much less frequently, the reverse appears to occur (i.e., a PA recurs as an apparent PMA). Whether the latter may occur due to overgrowth of the “loose” component (which often contains tumor cells with a higher overall proliferation index) or results from some sort of transformative event or simply represents sampling error in the initial biopsy is unclear at this time.
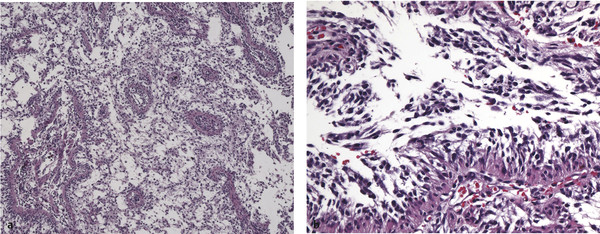
Fig. 43.2 Histologic appearance of pilomyxoid astrocytoma. (a) On low-power microscopy, the tumor is composed of scattered blood vessels within a purely microcystic background, lacking the characteristic biphasic appearance of pilocytic astrocytoma. (b) Characteristic of this tumor is a radial orientation of monomorphous tumor cells adhering to the blood vessel walls; this “angiocentricity” is better visualized at higher magnification. No Rosenthal fibers or eosinophilic granular bodies should be seen.
Diffuse Fibrillary Astrocytomas
DFAs (▶ Fig. 43.3) are WHO grade II tumors. The major (although variable) features that may help differentiate DFA from PA include more nuclear atypia and hyperchromasia, often less cellularity, and a more homogeneous cellular composition of fibrillary astrocytes (often without a clearly loose/microcystic component).29 The neoplastic cells definitely infiltrate brain. Mitoses should be relatively inconspicuous to absent for this to be classified as WHO grade II.30 Vascularity is much less prominent than in PA and PMA. DFA is much less common than PA and even PMA in the cerebellum. One helpful finding, which may be seen in smear preparations or sections, may be the increased presence of apoptosis or apoptotic bodies in DFA relative to PA (D.R.B., unpublished observation); otherwise, these two entities (PA and DFA) can be somewhat difficult to differentiate by routine histopathology alone, particularly with smaller samples. Another differentiating feature is that the piloid (long, thin, hair-like) processes typical of PA should not be a prominent feature in cytologic preparations of DFA, but this is not always a readily obvious difference. Most important may be the utilization of radiologic correlation to help differentiate these entities. WHO grade II DFA is nonenhancing and solid (not macrocystic), and it has blurry, infiltrative borders, in contrast to the typically enhancing, macrocystic, and discrete WHO grade I PA. Recently described molecular genetic markers may also be of further help in this differential. The “entity” of “diffuse” pilocytic astrocytoma is also described.14,31–33 Most practicing neuropathologists still consider this a WHO grade I neoplasm. This is a PA in which at least some significant portion infiltrates the cerebellum, especially if an infiltrative pattern of growth is noted even within the epicenter of the tumor; infiltration may be best demonstrated by the use of immunohistochemistry with antibodies to neurofilament protein (NFP), which demonstrate many NFP-positive cell processes/axons present within, and especially if present throughout, the specimen. In carefully examined gross total (and other large) resection specimens that are completely or nearly completely submitted for microscopic examination, at least a component of focally “diffuse” or infiltrating pattern is seen in many if not most cerebellar PAs. It is possible for such tumors, when suboptimally examined, to be mistakenly designated as “DFA” and thus “overgraded” as WHO grade II. Recent studies suggest that the biological behavior and overall prognosis for PA and for “diffuse” PA are similar, and that “diffuse” PA does not require any special clinical consideration or intervention in comparison with “classic” PA.14,32,33
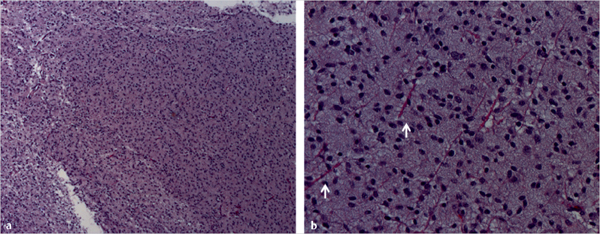
Fig. 43.3 Histologic appearance of diffuse fibrillary astrocytoma (World Health Organization grade II). (a) Low-power microscopy shows a homogeneous cellular pattern, with moderate hypercellularity and minimal pleomorphism and nuclear atypia. At higher magnification (b), it is possible to better observe a background composed of normal (pink) axonal processes (white arrows) demonstrating tumor infiltration into normal tissue. Mitotic activity is not seen.
Diffuse Anaplastic Astrocytoma and Glioblastoma Multiforme
These tumors (▶ Fig. 43.4) are both generally characterized by increased nuclear pleomorphism, hyperchromasia, and increased mitotic activity. Microvascular endothelial proliferation (multilayered “endothelium”) or pseudopalisading necrosis is pathognomonic for GBM.13,34 Bona fide “diffuse” (fibrillary, infiltrating) astrocytomas of the cerebellum, although infrequent overall, are more often of a higher grade (WHO grade III or IV rather than WHO grade II).14,32
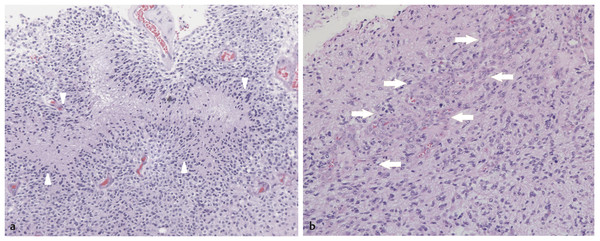
Fig. 43.4 Histologic appearance of glioblastoma multiforme. (a) At low magnification, it is possible to observe the pronounced hypercellularity, nuclear pleomorphism, and characteristic pseudopalisading of viable tumor cells about an area of tumor necrosis (white arrowheads). (b) A focus of vascular endothelial proliferation (white arrows) is causing nearly complete obliteration of blood vessel lumina; this finding (like pseudopalisading necrosis) is pathognomonic for glioblastoma multiforme (differentiating it from anaplastic astrocytoma).
43.3 Molecular Biology
Because of the frequently benign course of PA after surgical resection, its molecular signatures have not been studied as extensively as those of medulloblastoma, a tumor associated with a much worse prognosis and in need of better alternative therapies. However, the recent notions that not all pediatric cerebellar astrocytomas behave in the same way, that tumor recurrence does happen, and that adjuvant treatments may have to be considered in certain cases have stimulated the need for a deeper understanding of tumor biology.
Pediatric astrocytomas are molecularly different from the other astrocytic tumors that occur in adults. At the cytogenetic level, the karyotype is normal in at least 50 to 70% of cases35,36 and in up to 80% when only cerebellar astrocytomas are considered.37 Although adult astrocytomas are often characterized by loss of the short arm of chromosome 17 (17p) and long arm of chromosome 10 (10q), pediatric astrocytomas do not display a consistent karyotypic pattern,38 with the exception of the frequent duplication of chromosome 7q, which has been described in the majority of pilocytic astrocytomas and in 50% of diffuse fibrillary astrocytomas in children. Gain of 7q copy number results in gain of function of the gene BRAF.39 Unlike in adult astrocytomas, p53 mutation and loss of CDKN2A (p16) are not common findings in pediatric astrocytomas,40,41 and EGFR amplification is virtually absent.38,42
Among the molecular features that most consistently characterize PAs is the gain of function, by gene duplication, of BRAF, a serine/threonine protein kinase that, in turn, activates the MAPK/ERK cascade, leading to cellular proliferation.43 A recent analysis of 49 specimens of PA revealed duplication of the BRAF gene in 41 cases (83%), and this was twice as common in cerebellar as supratentorial tumors.44 As a proof of the connection to tumor pathogenesis, upregulation of BRAF expression in mouse neuroprogenitor cells induced the formation of intracranial PAs in vivo in 91% of cases.45 In addition, overexpression of BRAF in human neural stem cells induced an initial increase in clonogenic ability and proliferation, which was then followed by progressive cell senescence mediated by activation of p16. This phenomenon, known as oncogene-induced senescence (OIS), is characterized by a paradoxical decrease in cell growth or even total growth arrest initiated by oncogene induction, and it is triggered by the activation of tumor suppressor genes in response to abnormal proliferative stimuli. This observation may explain the often indolent behavior of the majority of PAs and low-grade pediatric astrocytomas, whose cells, although transformed, are still kept under control by the retained expression of major tumor suppressors, including p53 and p16.46 This may explain the high percentage of apoptotic cells observed in these tumors,47 as the result of the opposed action of proliferative and inhibitory stimuli. Although the association of supratentorial PA with NF-1 has been elucidated, and loss of heterozygosity of NF1, the gene encoding the tumor suppressor protein neurofibromin, has been recognized as the hallmark of PAs associated with NF-1, NF1 mutation has not been observed for sporadic tumors,48 suggesting a different molecular origin. A genetic screen comparing NF-1–associated with sporadic PAs revealed 26 genes differentially expressed between the two groups. Of these, ALDH1L1 expression has been associated with the NF-1–associated histology as well as with the more indolent tumors in general. In particular, ALDH1L1 downregulation has been found in 89% of PMAs and PAs with atypical features.49
With regard to the cell of origin of pediatric astrocytomas, immunostaining for SOX2, a transcription factor associated with “cancer stem cells,” was reported positive in about 10% of cells in 42 of 45 PAs.44 This raises the question whether a PA stem cell exists, as demonstrated for medulloblastoma, glioblastoma,50 and diffuse pontine glioma.51 In their seminal work on brain tumor stem cells, Singh et al were able to isolate cells with clonogenic ability from operative explants of PA that were immunoreactive for nestin and CD133, two of the accepted markers for cellular stemness. However, there has been no validation, to date, that these cells would be able to recapitulate tumor formation in vivo, as has been done for other brain tumors.52
43.4 Clinical Presentation
Posterior fossa tumors in children mainly present with symptoms of increased ICP, regardless of histology. This is due to progressive mass effect leading to obstruction of the fourth ventricle with resulting hydrocephalus, which is present in up to 85% of patients.7,10,11,22,25 Headache and vomiting are the two most common symptoms at presentation10,22 and are usually more pronounced in the morning because of recumbent position and increased cerebral blood volume secondary to higher PaCO2 .
The presentation also depends on the child’s age; in patients younger than 3 years, macrocrania and motor delay are common,12 whereas in older children, cerebellar signs are prominent and depend on the tumor location. Different from medulloblastoma and ependymoma, which present most commonly with truncal and gait ataxia due to their midline position, astrocytomas frequently present with limb ataxia that is secondary to hemispheric involvement. Papilledema, nystagmus, and diplopia are also frequently observed.7,10,11,22
The time to diagnosis is usually longer than for medulloblastoma because of the slower growth rate of astrocytomas; it ranges from weeks to several months,7,10 depending on tumor location. Typically, the symptomatology associated with vermian lesions progresses more quickly than that associated with hemispheric tumors. The sudden onset of symptomatology secondary to intratumoral bleeding has been described.53
43.5 Imaging
Computed tomography (CT) of the head with and without contrast has been historically considered the first diagnostic step for the evaluation of children presenting with symptoms suggestive of posterior fossa pathology (▶ Fig. 43.5). However, with the exception of the ability to differentiate between astrocytomas and medulloblastomas (decreased X-ray attenuation on unenhanced CT for the former, increased X-ray attenuation for the latter), CT is, in all other regards, inferior to magnetic resonance (MR) imaging of the brain for obtaining an accurate definition of tumor characteristics and anatomy. In our experience, limited MR imaging (axial T2-weighted sequence) serves as a screening examination in symptomatic patients. It can be performed as a safer, fast, and well-tolerated alternative to CT as an initial examination.54 If pathology is detected, formal MR imaging of the brain and spine will need to be obtained for surgical planning and prognostication.
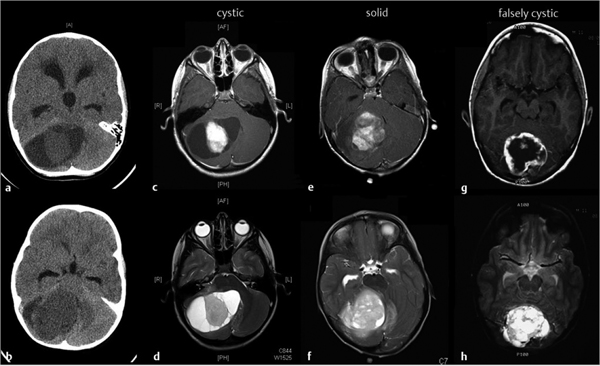
Fig. 43.5 Radiologic appearance of cerebellar pilocytic astrocytoma. Nonenhanced axial computed tomography shows either a hypo- or isodense nodule surrounded by signal, consistent with a fluid-filled cavity (a), or a slightly hypodense and heterogeneous mass in the case of a solid tumor (b). T1-weighted magnetic resonance imaging after the intravenous administration of gadolinium differentiates among “classic” cystic (c), solid (e), and falsely cystic (g) tumors. Neoplastic tissue enhances briskly upon contrast injection. In T2-weighted images, the solid component of the tumor (d,f,h) appears hyperintense to normal brain, a characteristic peculiar to pilocytic astrocytomas.
Regardless of imaging modality, cerebellar PAs appear as well-demarcated, often sizable lesions occupying the vermis (12.5 to 25%), hemispheres (36 to 40%), or concurrently the vermis and hemispheres (40%).55 Radiologically, astrocytomas have been described as presenting with three possible patterns (▶ Fig. 43.5):
Classic cystic astrocytoma presents as a usually large cystic lesion with an enhancing mural nodule but a nonenhancing cyst wall, which is considered nonneoplastic.
False cystic astrocytoma is characterized by the presence of a cystic component within the tumor. The tumor and cyst wall show contrast enhancement. The wall is thicker than 2 to 3 mm and is considered neoplastic.
Solid astrocytoma is devoid of macroscopic cystic formation and commonly occurs in the vermis.7,10,56 Fibrillary astrocytomas usually present as solid masses with less defined borders, suggesting invasion. They most often do not enhance.
Fewer than 10% of PAs invade the brainstem, a behavior more commonly observed with tumors arising in the midline and abutting the fourth ventricle.11,57,58 However, the presence and extent of brain stem invasion are usually difficult to predict preoperatively.
On MR imaging, the cystic component of PAs, when present, has a lower T1 signal and a higher T2 signal than that of brain. The mural nodule exhibits increased signal compared with brain on T1 images and enhances brightly and uniformly upon intravenous gadolinium administration. The extent of contrast enhancement of the cyst wall has been considered predictive of its nature; thick enhancement suggests a neoplastic cyst wall, whereas absent or limited enhancement (usually < 2 mm in thickness) is more consistent with reactive glial tissue.59 However, several authors failed to confirm a strong correlation between radiologic appearance and histology.22,60 Solid tumors usually enhance briskly, but up to 30% of lesions can display areas of decreased or absent enhancement.7 Particularly useful for the differentiation between astrocytomas and medulloblastomas are T2 images; the solid component of an astrocytoma consistently exhibits increased signal compared with brain, whereas a medulloblastoma exhibits similar or decreased signal compared with brain.61 Recently implemented apparent diffusion coefficient (ADC) maps have shown a specificity of 100% in differentiating cerebellar astrocytomas (higher signal intensity) from medulloblastomas (lower signal intensity).62 Peritumoral edema is rarely present and does not correlate with prognosis. Intratumoral calcifications are detectable in 25% of cases of PA.63
43.6 Treatment
Cerebellar astrocytoma is a surgical entity, and the primary treatment of cerebellar astrocytomas is surgical resection, with the goal of obtaining total tumor removal. In fact, extent of resection is the main predictor of recurrence and survival.
43.6.1 Preoperative Management
As discussed, children are usually diagnosed when already symptomatic secondary to increased ICP. Because of the potential danger of progressive hydrocephalus, surgical intervention should be planned in an urgent or semiurgent fashion. In those cases in which obtundation or cardiorespiratory instability is present, immediate intervention with placement of an external ventricular drain is mandatory. In these cases, maximum care needs to be paid to avoid excessive drainage, which can result in upward herniation. The preoperative placement of an external ventricular drain in nonemergent cases is matter of surgeon preference, but in our practice we prefer to avoid it and to start dexamethasone (0.25 to 0.5 mg/kg per day), which usually relieves symptoms in 6 to 12 hours.
43.6.2 Surgical Approach
Positioning is dictated by tumor location and the patient’s age. Usually, children younger than 2 years are placed prone with their head resting in a padded pediatric head holder. For older patients, rigid pin fixation is preferred, to increase stability and provide support for retracting devices.
For midline lesions, a vertical skin incision is performed from 1 cm above the external occipital protuberance to the upper cervical spine. An electric knife is then used to separate the posterior muscles of the neck. At this stage, to limit blood loss, care should be taken to cut through the ligamentous midline. Usually, the posterior arch of C1 is exposed but not removed. A suboccipital craniotomy is then performed, just below the transverse sinus and including the opening of the posterior rim of the foramen magnum. The dura is then opened in a Y pattern. It is important, at this stage, to be aware that, particularly in young children, the dura covering the posterior fossa is highly vascularized, so meticulous hemostasis is imperative. Depending on the size of the tumor, the mass effect exerted on the surrounding cerebellum may be substantial. In such cases, opening the cisterna magna is a useful strategy to drain some CSF and decrease ICP. Alternatively, for cystic tumors, a brain needle can be inserted through the dura into the cyst with subsequent aspiration of cyst fluid to achieve decompression.
For vermian lesions, the hemispheres are gently retracted bilaterally until the vermis is exposed and the tumor is visualized. Intraoperative ultrasound can aid in locating smaller tumors. A plane between PAs and surrounding cerebellum is usually well delineated. In order to delineate the anterior margin of the tumor, the fourth ventricle needs to be entered through the foramen of Magendie, so that, by insertion of a cottonoid, the floor of the fourth ventricle is protected during resection. The tumor is then resected piecemeal by aspiration and cautious coagulation. For large, solid lesions, an ultrasonic aspirator (e.g., Cavitron Ultrasonic Aspirator) can be employed. At the final stage, it is important to decide whether the cyst wall is neoplastic or reactive. To this end, when in doubt, multiple biopsies of the wall for frozen histology are recommended. It is preferable to leave the cyst wall in situ when it is not neoplastic.
For lesions located predominantly in the hemispheric region, a retrosigmoid approach may be considered. This has the advantage of sparing the midline structures of the cerebellum, thus preventing possible surgical complications, including cerebellar mutism.
For DFAs, the approach is the same, although the interface between normal tissue and tumor is less well defined. Surgical adjuvants, such as intraoperative frameless stereotaxy and intraoperative MR imaging, may provide useful guidance toward achieving the aim of a gross total resection (GTR).
In closure, the dura should be approximated in a watertight fashion, with the help, if needed, of an appropriate dural substitute. Dural closure is not essential but is advised to decrease the risks for chemical meningitis. Similarly, replacing the occipital bone is advised to decrease the risk for pseudomeningocele formation and to facilitate reoperation in case of tumor recurrence. Finally, it is imperative to meticulously approximate the muscle, fascia, and superficial layers to prevent CSF leakage and pseudomeningocele formation.
43.6.3 Surgical Complications
Postoperative mortality after posterior fossa tumor resection has been reported in the 1.5 to 5% range and is substantially decreased since the advent of modern microsurgical techniques.9,11,22 Common causes of mortality are surgical site hemorrhage, brain edema, and respiratory compromise secondary to brainstem violation.9 Common complications include CSF leakage, pseudomeningocele, meningitis (either infectious or aseptic), cranial nerve palsy, and ataxia.6,10,22 Cerebellar mutism is a unique consequence of posterior fossa surgery in children. Observed in 11 to 29% of patients, it is more commonly seen after the resection of vermian lesions, which explains why it is almost exclusively observed after medulloblastoma resection.64 Brainstem involvement by the tumor may also be a significant risk factor for this complication.65 Characteristically, it presents 1 to 6 days after an otherwise uneventful surgery with marked speech delay that is usually of limited duration (1 day to 4 months; average, 6 to 7 weeks). It can also be associated with ataxia, cranial nerve palsy, and emotional lability (the so-called posterior fossa syndrome). Recovery is spontaneous but frequently incomplete, as up to 68% of patients will have persistently impaired speech fluency and dysarthria 1 year after the resolution of mutism. Preoperative evidence of speech abnormalities has been found to be a positive predictor of postoperative mutism.66 The pathophysiology of cerebellar mutism is unknown, but most authors agree that it derives from bilateral perturbation of the dentate–thalamocortical pathways with resulting interruption of excitatory stimuli from the cerebellum to supratentorial speech centers. The delayed onset has been explained as resulting from postsurgical hypoperfusion,67 edema,68 transient deregulation of neurotransmitter release,69 or axonal injury caused by sudden release of the tumor mass effect.70
43.6.4 Postoperative Management
Intraoperative MR imaging at the end of tumor resection may be used to verify the extent of resection. Otherwise, MR imaging of the brain with and without intravenous gadolinium injection should be obtained within 48 hours of surgery to assess the extent of resection. In this time window, the presence of enhancement is strongly suggestive of residual disease. Depending on the MR imaging findings, the decisional algorithm is as follows: If a GTR has been obtained, no other treatment is indicated, and only follow-up imaging is recommended. There is no consensus, however, on how long the follow-up should be; because recurrence in the setting of GTR is a rare event, some authors have recommended a short follow-up schedule, suggesting only a single imaging session 6 months postoperatively.71 Contrarily, because of the indolent nature of these tumors, 8 to 10 years has been considered a reasonable period of follow-up by neuroimaging,72 although most commonly recurrence or progression happens within the first 5 years after initial treatment.73 If only subtotal resection has been achieved, early reoperation should be considered in cases in which it is deemed safe and feasible because of the survival advantage associated with GTR.74
If nonresectable, residual low-grade tumor is present, expectant management with frequent imaging is currently employed at most centers. This is justified by the unpredictable behavior of residual tumors, which not infrequently have been shown to remain stable or even regress at long-term follow-up. In a series of 168 benign cerebellar astrocytomas, Pencalet et al found that 42% of patients who had incomplete resection experienced recurrence, versus 5.6% of patients with initial GTR.22 In another study, of 14 children with residual tumor, 5 had progression, 5 had tumor regression, and the remainder had stable disease at 5 years.73 Gunny et al reported similar results, showing tumor regression in 45% of their patients during a follow-up period of longer than 6 years.75
The majority of patients with cerebellar astrocytoma show resolution of the hydrocephalus after tumor resection, although up to 15% will need CSF diversion for progressive hydrocephalus.10,76 In comparison, almost half of patients with medulloblastoma require shunt placement.76 Other than tumor histology, severe hydrocephalus, the presence of papilledema, and age younger than 2 years have been found to be significant predictors of shunt dependency.77 In patients who meet one or more of these criteria, frequent radiologic and clinical assessment is recommended. Indications for CSF shunting include evidence of enlarging ventricles, progressive symptomatology, and persistent CSF leak. Children who receive preoperative ventricular drainage have an increased likelihood to remain shunt-dependent postoperatively.78
43.6.5 Adjuvant Therapy
Patients whose tumor is completely resected do not need any further treatment. Chemotherapy and radiotherapy are usually considered in cases in which recurrence ensues or progression of residual disease is observed, if reoperation is not a possibility. Some authors have suggested that repeated resection should be attempted twice before radiotherapy is implemented in these patients.4 Others, particularly in light of recent results obtained with focal irradiation, suggest that noninvasive treatment should be favored to decrease surgical risks.
Upfront conventional radiotherapy to incompletely resected grade II gliomas failed to show any survival benefit compared with irradiation at tumor recurrence in a retrospective analysis of 90 patients younger than 20 years.79 Also, although usually associated with an excellent tumor response rate, conventional radiation has not been demonstrated to prolong survival.73,80,81 For these reasons, the consensus has been to reserve irradiation for relapsing low-grade astrocytomas and for higher-grade astrocytomas and GBM.7,13
More recent series describing results obtained with stereotactic radiosurgery have shown much more encouraging results; 50 patients with a diagnosis of PA who received stereotactic radiosurgery after partial resection or tumor recurrence had a 71% PFS at 5 years and 97.4% survival at 10 years. Small (< 8 cm3) size and solid rather than cystic tumor appeared to be the strongest variables for tumor response.82 In another study, 9 of 9 pediatric patients with residual cerebellar PA were progression-free at 5 years after treatment with conformal radiotherapy.83 Finally, in a Phase II trial enrolling patients with low-grade astrocytomas treated with conformal radiotherapy, Merchant et al reported 5- and 10-year PFS rates of of 87% and 74%, respectively, with a 10-year survival of 95%. Importantly, in the same series, the authors reported that cognitive impairment occurred only in children younger than 5 years. Other long-term side effects were hearing loss (4.9%), endocrinopathies, and vasculopathies (4.8%).84
Chemotherapy remains the front-line adjuvant therapy in infants and young children to defer radiotherapy and its deleterious effect on brain development and cognition. The combination of vincristine and carboplatin demonstrated tumor reduction in 56% of low-grade astrocytomas and a PFS rate at 3 years of 68%.85 Similarly, a tumor response in 70% of cases and a 3-year PFS rate of 78% was reported with a combination of cisplatin and etoposide.86 For tumors progressing despite prior chemotherapy, temozolomide has been shown to be a promising option, resulting in a 54% response rate and a 49% PFS rate at 2 years.87
43.7 Outcome
43.7.1 Prognostic Factors
The only two universally accepted and statistically validated factors associated with survival in patients with cerebellar astrocytomas are (1) low versus high grade and, in the case of low-grade tumors, (2) extent of tumor resection. Gender and age at diagnosis do not correlate with survival.7 There is still debate regarding whether low-grade diffuse astrocytomas are associated with worse outcome than PAs; Smoots et al found a statistically significant association of WHO grade II tumors with incidence of residual tumor in the brainstem and, consequently, with poor survival.88 This was in agreement with other series, published by Sgouros et al89 and Desai et al.11 More recently, results from a series of 200 patients failed to show a significant prognostic value for tumor histology.9 Size of the tumor, midline location, and solid appearance have all been associated with worse functional outcome.9
43.7.2 Leptomeningeal Dissemination
Fifty-eight cases of disseminated PA have been reported in the literature. The primary tumor was located in the cerebellum in 33% of cases, whereas the majority of cases originated from optic–hypothalamic lesions. PFS rates at 5 years ranged from 50 to 60% with a combination of surgery, radiation therapy, and chemotherapy. Craniospinal irradiation does not seem to be superior to focal radiotherapy and chemotherapy for disease control.18
43.7.3 High-Grade Gliomas
Malignant cerebellar astrocytomas are rare. When they arise “de novo,” they more frequently affect children younger than 3 years.12 Alternatively, they may result from malignant transformation of a prior low-grade tumor; malignant transformation of PA has been rarely reported, and in most such cases, it occurred after tumor irradiation.90,91 The time window between initial diagnosis and evidence of transformation ranged from 2 to 52 years.90 There are 25 cases of primary cerebellar GBM reported in the literature and 8 secondary cases of GBM resulting from malignant transformation of lower-grade tumors,13 of which 2 were PAs.90 Although symptoms at presentation are similar to those of lower-grade tumors, symptom progression is usually faster, and the diagnosis is made within a few weeks. A tendency for early leptomeningeal spread has also been reported.13 Despite aggressive surgical resection and adjuvant therapy, reported mean survival is 10 to 15 months.13,92
43.8 Recurrence/Progression
After GTR, recurrence has been reported in 0 to 6% of patients with low-grade astrocytomas,22,71–73,93 with a mean interval of 59 months after surgery.72 Following incomplete resection, the rate of tumor progression reaches 50% of cases, and progression usually ensues in the first 3 to 4 years.72,73 In the majority of cases, recurrence is diagnosed radiologically rather than clinically, justifying the implementation of long-term radiologic follow-up for patients with incompletely resected tumors.94 A recent series reported that 96% of the total recurrences/progressions happened within 8 years from initial surgery, and the authors concluded that radiologic follow-up should thus be extended to that interval.72 Although still debated, there does not seem to be a significant difference between recurrence or progression rates for PAs and for diffuse astrocytomas.72,94 In case of recurrence, reoperation is strongly recommended because it is associated with an excellent survival if GTR can be achieved7,93–95). When GTR is not a feasible option, subtotal resection followed by adjuvant chemotherapy and/or radiotherapy/radiosurgery is appropriate.
43.9 Survival
Patients in whom complete tumor resection is achieved have an excellent prognosis, with 10-year survival rates ranging from 90 to 100%.7,72,89 On the other hand, subtotal resection is associated with a higher likelihood of tumor progression, resulting in 10-year survival rates as low as 50% in some series,89,96 although other authors report more favorable outcomes, with up to 85% patients alive at 10-year follow-up.7 The mean survival of patients with GBM is 10 to 15 months.13
43.10 Functional Outcome
Up to 60% of patients treated for cerebellar PA and followed for a period of 8 years showed permanent neurologic deficits, mainly disequilibrium, strabismus, and ataxia. Furthermore, behavioral disorders, such as irritability and tearfulness, were present in more than 50%.97,98 However, in the majority of cases, the deficits did not prevent independent functioning and a high school education.97
43.11 Conclusion
Cerebellar astrocytomas are a heterogeneous group of tumors of childhood ranging from completely benign to extremely aggressive lesions. Low-grade tumors (WHO grades I and II) are the most frequent and are generally associated with a very good prognosis. Within this group, there is no agreement on whether PA carries a better prognosis than DFA, but evidence suggests that as long as complete resection is obtained, both have a very benign course, and cure can be very often achieved. The opposite is true for higher-grade tumors. To date, histology and extent of tumor resection are the only two validated predictors of PFS and overall survival; in light of this, the treatment plan for cerebellar tumors should always entail an aggressive approach aimed at complete resection (when this can be achieved with no or minor neurologic sequelae), and then further actions dictated by tumor grading and/or the presence of residual tumor. Patients with completely resected low-grade lesions should be followed by serial imaging. Second surgical resection should be considered for patients with residual tumor or recurrence. If this is not feasible, recent data support a role for stereotactic radiosurgery for local tumor control and extended PFS. Radiation is usually reserved for children older than 5 years of age to limit damage to the growing central nervous system. For younger children, chemotherapy is used. There are no indications to irradiate residual tumor before evidence of regrowth because spontaneous regression has been occasionally described. On the other hand, higher-grade tumors should receive maximum therapy, consisting of chemotherapy and irradiation following surgical resection.
Pearls
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


