Cerebrovascular Diseases: Introduction
Among all the neurologic diseases of adult life, stroke ranks first in frequency and importance. The common mode of expression of stroke is a relatively sudden occurrence of a focal neurologic deficit. Strokes are broadly categorized as ischemic or hemorrhagic. Ischemic stroke is due to occlusion of a cerebral blood vessel and causes cerebral infarction. The resultant neurologic syndrome corresponds to a portion of the brain that is supplied by one or more cerebral vessels. Knowledge of the stroke syndromes, the signs and symptoms that correspond to the region of brain that is supplied by each vessel, allows a degree of precision in determining the particular vessel that is occluded and from the temporal evolution of the syndrome, the underlying cause of vascular occlusion can be deduced.
Ischemic strokes are classified by the underlying cause of the vascular occlusion. One of three main processes is usually operative: (i) atherosclerosis with superimposed thrombosis affecting large cerebral or extracerebral blood vessels, (ii) cerebral embolism, and (iii) occlusion of small cerebral vessels within the parenchyma of the brain. There are many other pathologic processes that lead to ischemic brain damage, not all associated with occlusion of cerebral vessels, including arterial dissection, inflammatory conditions such as vasculitis, thrombosis of cerebral veins and dural sinuses, in situ thrombosis of large or small cerebral vessels due to hypercoagulable conditions, vasospasm from any of several mechanisms, unusual types of embolic materials such as fat, tumor, cholesterol, and several unique diseases that involve the cerebral vasculature (see further on). Closely allied with ischemic strokes is the transient ischemic attack (TIA), a temporary neurologic deficit caused by a cerebrovascular disease that leaves no clinical or imaging trace. The causes of stroke are so numerous that the listing given in Table 34-1 offers only a guide to the remainder of this chapter. As helpful is knowledge of the major causes of stroke by each epoch of age, particularly in childhood and young adults, a subject taken up in a later section and summarized in Table 34-2.
|
|
The second broad category consists of hemorrhage, which occurs either within the substance of the brain, intracerebral hemorrhage, or contained within the subarachnoid spaces and ventricular system, subarachnoid hemorrhage. The causes of the first category are numerous and include chronic hypertension, coagulopathies that arise endogenously or as a result of anticoagulant medications, vascular malformations of the brain, cranial trauma, and hemorrhage that occurs within the area of an ischemic stroke. Subarachnoid hemorrhage has fewer fundamental causes, the most common being the rupture of a developmental aneurysm arising from the vessels of the circle of Willis, but also includes cerebral trauma and arteriovenous malformations, and rarer processes.
The Clinical Stroke Syndrome
There are all gradations of severity, but in all forms of stroke the essential feature is abruptness with which the neurologic deficit develops—usually a matter of seconds that stamps the disorder as vascular. In its most severe form, the patient with a stroke becomes hemiplegic or even comatose—an event so dramatic that it had in the past been given vivid designations: apoplexy, cerebrovascular accident (CVA), or shock (colloquial). However, stroke is the preferred term.
In its mildest form, a stroke may consist of a trivial and transient neurologic disorder insufficient for the patient even to seek medical attention. Most embolic strokes occur suddenly and the deficit reaches its peak almost at once. Thrombotic strokes tend to evolve somewhat more slowly over a period of minutes or hours and occasionally days; in the latter case, the stroke usually progresses in a saltatory fashion, i.e., in a series of steps rather than smoothly. In cerebral hemorrhage, also abrupt in onset, the deficit may be virtually static or steadily progressive over a period of minutes or hours, while subarachnoid hemorrhage is almost instantaneous. It follows that gradual downhill course over a period of several days or weeks will usually be traced to a nonvascular disease. There are, however, many exceptions, such as the additive effects of multiple vascular occlusions and the progression that is caused by secondary brain edema surrounding large infarctions and cerebral hemorrhages. At the other extreme is rapid regression of a focal stroke syndrome that reverses itself entirely and dramatically over a period of minutes or up to an hour; this defines the TIA.
The second essential feature of stroke is its focal signature. The neurologic deficit reflects both the location and the size of the infarct or hemorrhage. Hemiplegia stands as the most typical sign of cerebrovascular diseases, whether in the cerebral hemisphere or brainstem, but there are many other manifestations, occurring in recognizable combinations. These include paralysis, numbness, and sensory deficits of many types on one side of the body, aphasia, visual field defects, diplopia, dizziness, dysarthria, and so forth. The neurovascular syndromes enable the physician to localize the lesion—sometimes so precisely that even the affected arterial branch can be specified—and to indicate whether the lesion is an infarct or a hemorrhage. These syndromes are described in the sections that follow. This group of diseases has also provided the most instructive approach to localization in neurology. As our colleague C.M. Fisher aptly remarked, neurology is learned “stroke by stroke.” Also, the focal ischemic lesion has divulged some of our most important ideas about the function of the human brain.
The analysis of a stroke involves several steps. First, the clinician must determine whether the event is a stroke rather than some other process that may have a similar sudden onset, such as migraine, seizure, or syncope. Second, if the event is considered likely to be a stroke or TIA, then the pathophysiology must be ascertained (e.g., cerebral embolism from the heart or a proximal artery, large vessel atherothrombotic occlusion, venous occlusive disease). Third, acute treatment (e.g., tissue plasminogen activator) is initiated, if appropriate. Fourth, a plan for the prevention of future strokes is undertaken.
In the last decades, extraordinary imaging technology has been introduced that allow the physician to make physiologic distinctions among normal, ischemic, and infarcted brain tissue. This approach to stroke will likely guide the next generation of treatments and has already had a pronounced impact on the direction of research in the field. Salvageable brain tissue in the acute phase of stroke can be delineated by these methods. To identify such ischemic but not yet infarcted tissue is a major goal of modern acute stroke medicine. In particular, diffusion-weighted magnetic resonance imaging has already altered the understanding and management of stroke patients.
The introduction of effective treatments for acute stroke has led to greater dependence on these sophisticated imaging techniques, but the authors believe it remains essential for the neurologist to understand the details of the cerebral vascular anatomy and the corresponding stroke syndromes for several reasons. Imaging techniques, though increasingly accurate, are not perfect. In cases in which the imaging does not reveal a stroke, the clinician remains dependent on careful history and neurologic examination. Furthermore, in many parts of the world, imaging techniques are unavailable at the pace necessary to initiate acute treatment. Finally, understanding the detailed anatomy helps the neurologist understand how the nervous system functions, lessons which are applicable to many other categories of illness other than stroke.
Despite these valuable imaging and therapeutic advances in stroke neurology, three points should be made. First, all physicians have a role to play in the prevention of stroke by encouraging the reduction of risk factors, such as hypertension, smoking, and hyperlipidemia and the identification of signs of potential impending stroke, such as transient ischemic attacks, atrial fibrillation, and carotid artery stenosis. Second, careful clinical evaluation integrated with the newer testing methods still provides the most powerful approach to this category of disease. Finally, there has been a departure from the methodical clinicopathologic studies that have been the foundation of our understanding of cerebrovascular disease. Increasingly, randomized trials involving several hundred and even thousands of patients and conducted simultaneously in dozens of institutions have come to dominate investigative activity in this field. These multicenter trials have yielded highly valuable information about the treatment of a variety of cerebrovascular disorders, both symptomatic and asymptomatic. However, this approach suffers from a number of inherent limitations, the most important of which is that the homogenized data derived from an aggregate of patients is difficult to apply to a specific case at hand or the data is not available to resolve each patient’s particular problem. Most large studies show only modest or marginal differences between treated and control groups and correspondingly give guidance in large populations. These multicenter studies will be critically appraised at appropriate points in the ensuing discussion.
The diagnosis of a vascular lesion therefore rests essentially on recognition of the stroke syndrome; without evidence of this, the diagnosis must always be in doubt. The three criteria by which the stroke is identified should be reemphasized: (1) the temporal profile of the clinical syndrome, (2) evidence of focal brain disease, and (3) the clinical setting. Definition of the temporal profile requires a clear history of the premonitory phenomena, the mode of onset, and the evolution of the neurologic disturbance in relation to the patient’s medical status. Here, an inadequate history is the most frequent cause of diagnostic error. If these data are lacking, the stroke profile may still be determined by extending the period of observation for a few days or weeks, thus invoking the clinical rule that the physician’s best diagnostic tool is a second and third examination. The first distinction is to separate ischemic from hemorrhagic stroke; features that are characteristic of the latter such as headache and vomiting at the onset, rapid progression to coma, and sever hypertension are emphasized in the later section on cerebral hemorrhage. Often, however, the distinction is not so clear because sudden onset of a focal neurologic problem is the core syndrome of both processes.
There are few categories of neurologic disease whose temporal profile mimics that of the cerebrovascular disorders. Migraine may do so, but the history usually provides the diagnosis. A seizure may be followed by a prolonged focal deficit (Todd paralysis) but is rarely the initial event in a stroke; the setting in which these symptoms occur and their subsequent course clarify the clinical situation. Tumor, infection, inflammation, degeneration, and nutritional deficiency are unlikely to manifest themselves precipitously, although rarely a primary or metastatic brain tumor produces a focal deficit of abrupt onset (see later). Trauma, of course, occurs abruptly but usually offers no problem in diagnosis. In multiple sclerosis and other demyelinative diseases, there may be an abrupt onset or exacerbation of symptoms, but for the most part they occur in a different age group and clinical setting. Conversely, a stroke-like onset of cerebral symptoms in a young adult should always raise a suspicion of demyelinative disease. A stroke developing over a period of several days usually progresses in a stepwise fashion, increments of deficit being added abruptly from time to time. A slow, gradual, downhill course over a period of 2 weeks or more indicates that the lesion is probably not vascular but rather neoplastic, demyelinative, infectious (abscess) or granulomatous, or a subdural hematoma.
In regard to the focal neurologic deficits of cerebrovascular disease, many of the nonvascular diseases may produce symptoms that are much the same, and the diagnosis cannot rest solely on this aspect of the clinical picture. Nonetheless, specific patterns of neurologic signs are so highly characteristic of vascular occlusion—e.g., the lateral medullary syndrome—that they mark the disease as a stroke. Conversely, certain disturbances are hardly ever attributable to ischemic stroke—e.g., diabetes insipidus, fever, bitemporal hemianopia, parkinsonism, generalized myoclonus, repeated falls, and isolated cranial-nerve palsies—and their presence may be of help in excluding vascular disease. Finally, the diagnosis of cerebrovascular disease should always be made on positive data, not by exclusion.
A few conditions are so often confused with cerebrovascular diseases that they merit further comment. Miscellaneous conditions occasionally taken to be a stroke are migraine; Bell’s palsy; Stokes-Adams syncopal attacks; a severe attack of labyrinthine vertigo; diabetic ophthalmoplegia; acute ulnar, radial, or peroneal palsy; embolism to a limb; and temporal arteritis associated with blindness all of which are discussed in later parts of this chapter.
A brain tumor, especially a rapidly growing glioblastoma or lymphoma, may produce a severe hemiplegia rapidly. Also, the neurologic deficit caused by cancer metastatic to the cerebrum may evolve rapidly, almost at a stroke-like pace. Moreover, in rare cases of brain tumor, a hemiplegia may be preceded by transitory episodes of neurologic deficit, indistinguishable from TIAs. The presence of the tumor and its effects on the cerebrum may make it difficult for the patient to articulate a clear history. A lack of detailed history may also be responsible for the opposite diagnostic error, i.e., mistaking a relatively slowly evolving stroke (usually caused by internal carotid artery or basilar occlusion) for a tumor. CT and MRI usually settle the problem. A brain abscess or inflammatory necrotic lesion—e.g., herpes encephalitis or toxoplasmosis—may also develop rapidly.
Contrariwise, certain manifestations of stroke may be incorrectly interpreted as evidence of some other neurologic disorder. Headache, at times severe, often occurs as a prodrome of a thrombotic stroke or subarachnoid hemorrhage; unless this is appreciated, a diagnosis of migraine may be made. Dizzy spells, vertigo, vomiting, or brief intermittent lapses of equilibrium as a result of vascular disease of the brainstem may be ascribed to vestibular neuritis, Ménière disease, Stokes-Adams syncope, or gastroenteritis. A detailed account of the attack will usually avert this error. A strikingly focal monoplegia of cerebral origin, causing only weakness of the hand or arm or foot-drop, is not infrequently misdiagnosed as a peripheral neuropathy or plexopathy.
Stroke, after heart disease and cancer, is the third most common cause of death in the United States. Every year there are in the United States approximately 700,000 cases of stroke—roughly 600,000 ischemic lesions and 100,000 hemorrhages, intracerebral or subarachnoid—with 175,000 fatalities from these causes combined. Since 1950, coincident with the introduction of effective treatment for hypertension, there has been a substantial reduction in the frequency of stroke. Both sexes have shared in the reduced incidence. During this period, the incidence of coronary artery disease and uncontrolled hypertension also fell significantly. By contrast, there has been no change in the frequency of aneurysmal rupture. In the last two decades, according to the American Heart Association, the mortality rate from stroke has declined by 12 percent, but the total number of strokes may again be rising. Stroke assumes importance both because of its high rate of mortality and the residual disability that it causes.
The burden of stroke has far wider implications when viewed from an international perspective. Cerebrovascular disease is estimated to account for 7.8 million deaths yearly throughout the world and represents about 13 percent of all causes of death. In developed countries, stroke mortality is only surpassed by cardiac ischemic diseases and close to equivalent to the cancers collectively (mainly lung cancer) in the most recent Global Burden of Disease study undertaken in 2004. Stroke remains among the five leading causes of death across every income group in most countries in the last comprehensive review by the World Health Organization in 2004. They cause significant physical, emotional, and cognitive disabilities among survivors, accounting for 3.6 percent of the total disability-adjusted life years (DALYs) and thus placing stroke within the 10 leading causes of disability irrespective of the development status of countries (see ).
This is an area of major public health importance in that several modifiable factors are known to increase the liability to stroke. The most important of these are hypertension, atrial fibrillation, diabetes mellitus, cigarette smoking, and hyperlipidemia. Others, such as systemic diseases associated with a hypercoagulable state and the use of contraceptives, also contribute, but only in special circumstances. Hypertension is also the most readily recognized factor in the genesis of primary intracerebral hemorrhage. It appears that the stroke-producing potential of hypertension is as much the product of heightened systolic pressure, as of diastolic pressure (Rabkin et al). The cooperative studies of the Veterans Administration (see Freis et al) and the report by Collins and associates (collating 14 randomized trials of antihypertensive drugs) convincingly demonstrated that the long-term control of hypertension decreased the incidence of both ischemic infarction and intracerebral hemorrhage. It has been found that simple measures such as the use of hydrochlorothiazide for blood pressure control may be, overall, the most effective. The presence of congestive heart failure and coronary atherosclerosis also increases the probability of stroke. As for embolic strokes, the most important risk factors are structural cardiac disease and arrhythmias, mostly atrial fibrillation, which increases the incidence of stroke about 6-fold, and by 18-fold if, as was common in the past, there is also rheumatic valvular disease.
Diabetes hastens the atherosclerotic process in both large and small arteries. Weinberger and colleagues and Roehmholdt and coworkers found diabetic patients to be twice as liable to stroke as age-matched nondiabetic groups. The importance of long-duration cigarette smoking in the development of carotid atherosclerosis has long been known and was quantitated by Ingall and colleagues. The interactions between diabetes and hypertension on the one hand, and intracerebral hemorrhage and atherothrombotic infarction on the other, as well as the association of cardiac disease and cerebral embolism, are considered further on in this chapter in relation to each of these categories of cerebrovascular disease. Numerous clinical trials have also shown a marked reduction in stroke incidence with the use of cholesterol-lowering drugs. As in the case of coronary artery disease, the level of low-density lipoprotein (LDL) cholesterol has the most impact on the incidence of stroke but elevated triglycerides may also confer risk. Subsidiary factors, such as low potassium intake and reduced serum levels of potassium, are associated with an increased stroke rate in several studies, including one in which we participated, but the mechanism of this effect is obscure (Green et al); a detrimental effect on blood pressure is possible. Public health measures designed to detect and reduce the aforementioned risk factors offer the most intelligent long-range approach to the prevention of cerebrovascular disease.
Finally, in keeping with the emerging field of genetic risk factors in human disease, several genetic loci have been found that putatively impart a risk of stroke in various populations. The largest of these, reported by Ikram and associates, has implicated a polymorphism on chromosome 12, encompassing several genes that have putative connections to vascular disease. However, other groups, such the International Stroke Genetics Consortium, were unable to confirm this. It seems likely that more refined definitions of stroke subtypes and careful genotyping of circumscribed populations will be necessary if genetic risk factors for stroke are to be be found that are not simply markers for vasculopathy, inducing diseases such as diabetes, hyperlipidemia, and hypertension.
Two causes of ischemic stroke stand out: atherosclerotic–thrombotic disease of the cerebral or extracerebral vessels, and cerebral embolism. An understanding of the biology of these two processes is essential for the analysis of the clinical, laboratory, and imaging features of stroke and its treatment. All other causes of vascular occlusion taken together, account for far fewer strokes. These are also important and they are accorded their own sections later in the chapter.
The evolution of clinical phenomena in cerebral thrombosis, both of large intracranial (basilar, carotid) or extracranial (carotid, vertebral) and small vessels (lacunes), is more variable than that of embolism and hemorrhage. In approximately half of patients the stroke is preceded by minor signs or one or more transient attacks of focal neurologic dysfunction, TIAs, discussed further on. These transient prodromal episodes may herald the oncoming vascular event caused by atherothrombotic stroke. Occasionally embolism is preceded by a transient neurologic disorder but TIAs are generally considered as more closely aligned with atherothrombotic stroke.
The thrombotic stroke syndrome develops in one of several ways. There may be a single episode, but typically the whole stroke evolves over a few minutes or hours or less. Characteristic is a “stuttering” or intermittent progression of neurologic deficits extending over several hours or a day or longer. This is a starkly different profile from the abrupt onset of stroke that characterizes the embolic mechanism discussed further on. In thrombosis, a partial stroke may occur and even recede temporarily for several hours, after which there is rapid progression to the completed deficit—or several fleeting episodes may be followed by a longer one and, hours or a day or two later, by a major stroke. Several parts of the body may be affected at once or only one part, such as a limb or one side of the face, the other parts becoming involved serially in step-like fashion until the stroke is fully developed. Sometimes the deficit is episodic; spells of weakness or involuntary movement of a hand or arm or dimness of vision, lasting 5 to 10 min, occur spontaneously or are brought on by standing or walking. Each of the partial attacks may reproduce the profile of the stroke in miniature. In other words, the principle of intermittency seems to characterize the thrombotic process from beginning to end.
Also characteristic of atherothrombotic events in many, but not all cases, is the occurrence of the stroke during sleep; the patient awakens paralyzed, either during the night or in the morning. Unaware of any difficulty, he may arise and fall helplessly to the floor with the first step. This is the story given by half of our patients with thrombotic strokes, as well as by a smaller number with embolic strokes.
Most deceptive are the few instances, in which the neurologic disorder evolves very gradually, over several days or longer (“slow stroke”). One’s first impulse is to make a diagnosis of brain tumor, abscess, or subdural hematoma. This error can usually be avoided by a careful analysis of the course of the illness, which will disclose an uneven, saltatory progression. There are also cases—and these are usually instances of pure motor hemiplegia—in which the evolution of a thrombotic stroke is evenly progressive over a period of days. It is likely also that the abrupt development of a thrombus on an atherosclerotic plaque in a distal cerebral vessel (beyond the circle of Willis) can also cause a fairly sudden or at least rapid evolution of stroke, but this is not characteristic.
Atheromatous plaques preferentially form at branching points and curves of the cerebral arteries. The most frequent sites are (1) in the internal carotid artery at its origin from the common carotid; (2) in the cervical part of the vertebral arteries and at their junction to form the basilar artery; (3) in the stem or at the main bifurcation of the middle cerebral arteries; (4) in the proximal posterior cerebral arteries as they wind around the midbrain; and (5) in the proximal anterior cerebral arteries as they pass anteriorly and curve over the corpus callosum. The last two sites are far less frequent than the first three. It is infrequent for the cerebral arteries to develop significant plaques beyond their first major branching after the circle of Willis. Also, it is unusual for the cerebellar and ophthalmic arteries to show atheromatous involvement. The common carotid and vertebral arteries at their origins from the aorta are additional frequent sites of atheromatous deposits, but because of abundant collateral arterial pathways, occlusions at these sites are less commonly associated with cerebral ischemia as discussed further on.
Atherothrombosis may cause cerebral infarction in several ways. The most obvious is that an occlusive plaque or a thrombus formed on a plaque occupies the lumen of a major intracerebral vessel, such as the middle cerebral artery, and stops flow to the areas of the brain supplied by the vessel. A variation of this mechanism is one of occlusion by atherosclerosis of a more proximal vessel, such as the distal carotid artery. This leads to infarction in the territory between major branches of the internal carotid circulation that are most susceptible to reduced blood flow—termed “watershed infarction.” Or, an atherothrombotic lesion in a proximal vessel may serve as the nidus for the formation of an embolus that manifests itself as a stroke in one of the territories of that vessel—called “artery-to-artery” embolism. The essential issue here is that it is almost always severe stenosis of a proximal internal carotid artery that produces either ischemic or embolic stroke; milder degrees are usually not implicated. A separate mechanism pertains when an atherosclerotic plaque in a large vessel of the circle of Willis occludes the orifices of small penetrating vessels, most often the lenticulostriate branches of the middle cerebral artery or the thalamostriate vessels of the posterior cerebral artery, and cause small, or more confluent, strokes deep in the brain.
Whether plaque rupture plays a role in vessel occlusion or thrombus formation, as it does in the coronary artery, is not clear. In the carotid artery, Hosseini and coworkers found evidence of intraplaque hemorrhage using special MRI techniques and found these changes to be predictive of stroke in the distal distribution of the artery. Previous work by Fisher and Ojemann, cited in the references, involving the serial sectioning of carotid plaques removed at surgery, suggested otherwise. It is clear that the more severe the focal atheromata, the more likely a thrombotic complication will occur. Whether the complexity of a carotid artery plaque with ulcerations is an important component of stroke risk, for example, by originating small emboli, is not entirely settled. Again, it is the high degree of stenosis, usually above 90 percent of the original lumen compromised, or a residual lumen of less than approximately 2 mm, of the carotid artery that is most likely to be associated with strokes in the distal territory of the vessel.
Atheromatous lesions may regress to some extent under the influence of diet and lipid-lowering drugs. Hennerici and colleagues followed a series of patients with carotid stenosis for a period of 18 months and observed spontaneous regression in nearly 20 percent of the lesions. In the large majority of cases, however, atherosclerosis is a progressive disease.
The hemostatic elements, both clotting factors and platelets, which produce a thrombus within a vessel are complex and have been the object of intense study (see Furie and Furie for a discussion of this field). However, just as in the case of coronary artery disease, it is often the development and enlargement of the thrombus that acts as the final element in cerebral vascular occlusion and an ischemic stroke. It seems plausible, although not adequately studied, that the temporal profile of atherothrombotic stroke reflects this accretion of clot in a vessel. These biologic mechanisms have bearing on the treatment and prevention of stroke.
This is the most common cause of ischemic strokes and of all the types of stroke, cerebral embolism develops most rapidly, “like a bolt out of the blue.” As a rule, the full-blown picture evolves within seconds, exemplifying most perfectly the idealized temporal profile of a stroke. Although the abruptness with which the stroke develops and the lack of prodromal symptoms point strongly to embolism, the diagnosis is based on the total clinical circumstances. Embolism always merits careful consideration in young persons, in whom atherosclerosis is less common. Only occasionally does the problem unfold more gradually, over many hours, with some fluctuation of symptoms. Possibly, in these cases the embolus initiates a propagating thrombotic process in the occluded vessel.
In most cases, the embolic material consists of a fragment that has broken away from a thrombus within the heart (“cardioembolic”). Somewhat less frequently, the source is intraarterial from the distal end of a thrombus within the lumen of an occluded or severely stenotic carotid or vertebral artery, or a clot that originates in the systemic venous system and passes through an aperture in the heart walls, or the origin of an embolus may be from large atheromatous plaques in the aorta. Thrombotic or infected material (endocarditis) that adheres to the aortic or mitral heart valves and breaks free are also well-appreciated sources of embolism, as are clots originating on prosthetic heart valves. Embolism caused by fat, tumor cells (atrial myxoma), fibrocartilage, amniotic fluid, or air enters into the differential diagnosis of stroke only in special circumstances.
The embolus usually becomes arrested at a bifurcation or other site of natural narrowing of the lumen of an intracranial vessel. The resultant infarction is pale, hemorrhagic, or mixed; hemorrhagic infarction nearly always indicates embolism (although venous occlusion can do the same). Any region of the brain may be affected, the territories of the middle cerebral artery, particularly the superior division, being most frequently involved. The two cerebral hemispheres are approximately equally affected. Large embolic clots can block large vessels (e.g., the carotid arteries in the neck or at their termination intracranially), while tiny fragments may reach vessels as small as 0.2 mm in diameter, usually with inconsequential effects. The embolic material may remain arrested and plug the lumen solidly, but more often it breaks into fragments that enter smaller vessels so that even careful pathologic examination fails to reveal their final location. In this instance, the clinical effects may abate. Because of the rapidity with which embolic occlusion develops, useful collateral influx does not become established. Thus, sparing of the brain territory distal to the site of occlusion is usually not as evident as in thrombosis that develops more slowly.
According to the Framingham Heart Study, patients with chronic atrial fibrillation are approximately six times more liable to stroke than an age-matched population with normal cardiac rhythm (Wolf et al, 1983) and the risk is considerably higher if there is also rheumatic valvular disease, now far less prevalent than in the past. Furthermore, the risk for stroke conferred by the presence of atrial fibrillation varies with age, being 1 percent per year in persons younger than age 65 years, and as high as 8 percent per year in those older than age 75 years with additional risk factors. These levels of risk are of prime importance in determining the potential benefit of chronic anticoagulation, as discussed later. Embolism may also occur in cases of paroxysmal atrial fibrillation or flutter and various studies have suggested that the risk of stroke is even greater than for the chronic arrhythmia. Even more vexing, intermittent and asymptomatic atrial fibrillation is difficult to detect except with long periods of rhythm monitoring. For example, in a study of patients with implanted pacemakers or defibrillators but not known to have atrial fibrillation by Healy and colleagues, a substantial number of atrial arrhythmias were uncovered and raised the risk of stroke fivefold. In related studies by Gladstone and coworkers and by Gaillard and colleagues, suggest that recording heart rhythm for longer periods with a loop monitor increases the rate of detection of episodic atrial fibrillation to approximately 15 percent, from approximately 3 percent with conventional Holter monitoring. Whether such long-term monitoring should be adopted in routine practice is not yet certain, but it is being increasingly added in the evaluation of “cryptogenic” stroke.
Several scoring systems have been developed to gauge the future likelihood of stroke from atrial fibrillation. The CHADS2 and related systems are shorthand methods to quantitate the risk factors that modulate risk for stroke in a patient with atrial fibrillation. A refinement of this system, CHA2DS2-VASc is purported to improve these predictions but the confidence intervals around the point estimates of predictive values in both scales are considerable and clinical judgment must be exercised in their use. This is reflected in part by the observation that the second score does not confer increased risk of stroke in a continuous fashion with each increase in score. The scores, subject to replacement by future ones, are given in Table 34-3. Furthermore, the goal of most of these systems is to make choices regarding warfarin or similar anticoagulation for the prevention of embolic stroke from the arrhythmia as discussed later in the chapter, or as pertinently to identify patients who have such a low risk of stroke that the risks of anticoagulation may not be justified. Epidemiologic and clinical aspects of the protective effects of anticoagulation have their own imprecisions.
CHADS2 | POINTS ASSIGNED FOR EACH ITEM | PREDICTED YEARLY STROKE RISK BY TOTAL SCORE |
|---|---|---|
Congestive heart failure | 1 | 0…. 1.9% |
Hypertension | 1 | 1….. 2.8% |
Stroke, TIA in past | 2 | 2….. 4.0% |
Vascular disease | 1 | 3… 5.9% |
Diabetes | 1 | 4… 8.5% |
Female | 1 | 5.. 12.5% |
Age | ||
<65 years | 0 | 4….. 8.5% |
65–74 years | 1 | 5….12.5% |
>75 years | 2 | 6….18.2% |
CHA2DS2-VASC | POINTS ASSIGNED FOR EACH ITEM | 0…. 0% |
Heart failure or ejection fraction <35% | 1 | 1…… 1.3% |
Hypertension | 1 | 2…… 2.2% |
Age <65 years | 0 | 3…… 3.2% |
66–74 years | 1 | |
>75 years | 2 | |
Previous stroke or TIA | 2 | 4…… 4.0% |
Diabetes | 1 | 5…….6.7% |
Coronary or peripheral vascular disease | 1 | 6…… 9.8% |
Female | 1 | 7….. 6.9% |
8….. 6.7% | ||
9….. 15.5% |
Mural thrombus deposited on the damaged endocardium overlying a myocardial infarct in the left ventricle, particularly if there is an aneurysmal sac, is an important source of cerebral emboli, as is a thrombus associated with severe mitral stenosis without atrial fibrillation, now a far less common circumstance than when rheumatic fever was prevalent. Emboli may occur in the first few weeks after an acute myocardial infarction but Loh and colleagues found that a lesser degree of risk persists for up to 5 years. Cardiac catheterization or surgery, especially valvuloplasty, may disseminate fragments from a thrombus or a calcified valve. Mitral and aortic valve prostheses are, as mentioned, additional important sources of embolism.
Another source of embolism is the carotid or vertebral artery, where clot forming on an ulcerated atheromatous plaque may be detached and carried to an intracranial branch (artery-to-artery embolism). A similar phenomenon occurs with arterial dissections, discussed in a later section, “Less-Common Causes of Ischemic Cerebrovascular Disease,” and sometimes with fibromuscular disease of the carotid or vertebral arteries.
Atheromatous plaques in the ascending aorta have been recognized to be a more frequent source of embolism than had been previously appreciated. Amarenco and colleagues reported that as many as 38 percent of a group of patients with no discernible cause for embolic stroke had echogenic atherosclerotic plaques in the aortic arch that were greater than 4 mm in thickness, a size found to be associated on a statistical basis with strokes. Disseminated cholesterol emboli from the aorta are known to occur in the cerebral circulation and may be dispersed to other organs as well; rarely, this is sufficiently severe to cause an encephalopathy and pleocytosis in the spinal fluid.
Also of interest are the symptoms caused by an embolus as it traverses a large vessel. This migrating or traveling embolus syndrome is most evident in cases of posterior cerebral artery occlusion, either from a cardiogenic source or from a thrombus in the proximal vertebral artery (“artery-to-artery” embolism; see Koroshetz and Ropper). Minutes or more before the hemianopia develops, the patient reports fleeting dizziness or vertigo, diplopia, or dysarthria, the result of transient occlusion of the origins of penetrating vessels as the clot material traverses the basilar artery. Small residual areas of infarction within the brainstem or cerebellum can be seen on MRI or found at autopsy, and some of the signs of brainstem infarction may persist. The basilar artery is singularly susceptible to this syndrome because the vertebral arteries are smaller in caliber than the basilar, allowing a clot to slowly traverse the larger vessel; furthermore, a clot in the basilar artery is prone to occlude the small orifices of arteries that supply blood to the brainstem.
Paradoxical embolism occurs when an abnormal communication exists between the right and left sides of the heart (particularly a patent foramen ovale [PFO]) or the alternative route of connection via a pulmonary arteriovenous fistula. Embolic material arising in the veins of the lower extremities or pelvis or elsewhere in the systemic venous circulation bypasses the pulmonary circulation and reaches the cerebral vessels. Pulmonary hypertension (often from previous pulmonary embolism) favors the occurrence of paradoxic embolism, but these strokes occur even in the absence of pulmonary hypertension. Several studies indicate that the presence of a small atrial septal aneurysm adjacent to the patient foramen increases the likelihood of stroke. In the series reported by Mas and colleagues (2001), patients ages 18 to 55 years who had a stroke were followed for 4 years; the risk of second stroke was 2 percent in those with a PFO alone and 15 percent among those with both a PFO and an atrial septal aneurysm (curiously, the risk among those with neither congenital abnormality was 4 percent—higher than for those with a PFO alone). This mechanism comes into play mainly in considering the causes of stroke in the younger patient, but Handke and colleagues published a series in which there was a slightly increased risk of stroke in patients who were older than age 55 and had PFO. It must be emphasized, however, that about one-third of patients in all age groups will be found to have a PFO, and anticoagulation or repair of these lesions in older patients with embolic stroke has not been shown to be beneficial (see further on for discussion of treatment of PFO). Subendocardial fibroelastosis, idiopathic myocardial hypertrophy, cardiac myxomas, and myocardial lesions of trichinosis are additional rare causes of embolism from a cardiac source. The vegetations of infective and noninfective (marantic) endocarditis give rise to several different lesions in the brain as described in Chap. 32.
Mitral valve prolapse, in the past considered a common source of emboli, especially in young patients, is no longer currently thought to be an important origin. The initial impetus for considering this abnormality as a source of embolus came from the study of Barnett and colleagues (1980) of a group of 60 patients who had TIAs or small strokes and were younger than 45 years of age; mitral prolapse was detected (by echocardiography and a characteristic midsystolic click) in 24 patients, but in only 5 of 60 age-matched controls. However, several subsequent large studies (Sandok and Giuliani; Jones et al) found that only a very small proportion of strokes in young patients could be attributed to prolapse; even then, the connection was inferred by the exclusion of other causes of stroke. Indeed, in a study using stringent criteria for the echocardiographic diagnosis of prolapse, Gilon and colleagues were unable to establish a relation to stroke. Usually, when valvular prolapse is associated with stroke, it is usually severe with ballooning of the valve and a propensity to accumulate clot behind the valve. Of interest, Rice and colleagues described a family with premature stroke in association with valve prolapse and a similar relationship has been reported in twins; the same may occur in Ehlers-Danlos disease.
The pulmonary veins are a potential, if infrequent, source of cerebral emboli, as reflected by the occurrence of cerebral abscesses in association with pulmonary infectious disease (and by the high incidence of cerebral deposits secondary to pulmonary carcinoma). In Osler-Weber-Rendu disease, pulmonary shunts serve as a conduit for emboli. A rare type of embolism follows thyroidectomy, where thrombosis in the stump of the superior thyroid artery extends proximally until a section of the clot, protruding into the lumen of the carotid artery, is carried into the cerebral circulation.
During cerebral arteriography, emboli may arise from the tip of the catheter, or manipulation of the catheter may dislodge atheromatous material from the aorta or carotid or vertebral arteries and account for some of the strokes during this procedure. Monitoring of the cerebral arteries by transcranial Doppler insonation has suggested that small emboli frequently arise during angiographic procedures. For example, a study by Bendszus and colleagues found that 23 of 100 consecutive patients had new cortical lesions shown on diffusion-weighted MRI just after cerebral arteriography. However, none of these patients was symptomatic, and with good technique, emboli from vascular catheters are infrequent.
Cerebral embolism of special type must always have occurred when secondary metastatic tumor is deposited in the brain but a mass of tumor cells is seldom large enough to occlude a cerebral artery and produce the picture of a stroke. Nevertheless, tumor embolism with stroke is known from cardiac myxoma and fibroelastoma, and occasionally with other tumors, even systemic ones; in some of these cases it is a thrombus in the primary lesion that offers a source of embolism. This syndrome must be distinguished from embolism caused by nonbacterial endocarditis that complicates malignant neoplasms (nonbacterial thrombotic endocarditis is discussed further on). This special source of cerebral embolism is a component of a hypercoagulable state that especially accompanies adenocarcinoma and cachexia.
Diffuse cerebral fat embolism is related to severe bone trauma. As a rule, the emboli are minute and widely dispersed, giving rise first to pulmonary symptoms and then to multiple dermal (anterior axillary fold and elsewhere) and cerebral petechial hemorrhages. Accordingly, the clinical picture is more of an encephalopathy and not strictly focal as it is in a stroke, although in some instances there may be focal features. Cerebral air embolism is a rare complication of abortion, scuba diving, or cranial, cervical, or thoracic operations involving large venous structures or venous catheter insertion; it was formerly encountered as a complication of pneumothorax therapy. Clinically, cerebral air embolism may be difficult to separate from the deficits following hypotension or hypoxia with which it frequently coexists. Hyperbaric treatment may be effective if instituted early.
Despite the large number of established sources of emboli, the point of origin cannot be determined in 20 to 30 percent or more of presumed embolic strokes. In such cases, emboli likely have originated from thrombi in the cardiac chambers but have left behind no residual clot and may be undetectable even by sophisticated methods, such as transesophageal echocardiography and newer MR techniques. Other cases may be a result of atheromatous material arising from the aorta or paradoxical embolism. If extensive evaluation fails to disclose the origin, the odds still favor a source in the left heart. Often, the diagnosis of cerebral embolism is made at autopsy without finding a source. In these cases, one presumes that the search for a thrombotic nidus may not have been sufficiently thorough and small thrombi in the atrial appendage, endocardium (between the papillary muscles of the heart), the aorta and its branches, or pulmonary veins may have been overlooked. Nevertheless, the source of embolic material is not revealed in a number of cases.
When brief ischemic attacks precede a stroke but disappear entirely, leaving no clinical or imaging trace of cerebral infarction, they almost always stamp the underlying process as atherothrombotic involving a large or small blood vessel. Transient ischemic attacks can reflect the involvement of virtually any cerebral artery: common or internal carotid; middle, posterior, or anterior cerebral; ophthalmic; vertebral, basilar, or cerebellar; or a penetrating branch to the internal capsule, thalamus, or brainstem (lacunar TIAs). Thus, they may present themselves as transient spells of hemiparesis, aphasia, numbness or tingling on one side of the body, dysarthria, diplopia, ataxia, obscuration of a visual field, or combinations thereof that replicate the stroke syndromes. TIAs may precede, accompany, or infrequently follow the development of a stroke, or they can occur by themselves without leading to a stroke—a fact that makes any form of therapy difficult to evaluate. Transient ischemic episodes must be distinguished from other brief neurologic attacks that are from seizures, migraine and its variants, transient global amnesia, syncope, vertigo from labyrinthine disease, and psychogenic episodes as emphasized further on. The differentiation of TIAs from other similar transient spells is not always straightforward and occupies considerable attention from neurologists; the implications of these distinctions have serious implications with regard to evaluation and treatment.
Although there is little doubt that TIAs are caused by transient focal ischemia, their mechanism is not fully understood. Current opinion holds that TIAs are brief, reversible episodes of focal ischemic brain disturbance without evidence of cerebral infarction. The consensus had in the past been that their duration should be less than 24 h, an idea introduced 40 years ago by a committee assigned to study the problem. It is more useful clinically to separate attacks that last only a few minutes (up to 1 h) and leave no permanent signs, from those of longer duration, which are almost invariably a result of embolism that shows evidence of infarction on imaging studies and therefore carry an entirely different connotation than briefer TIAs. Whether to redefine TIAs by the presence or absence of imaging abnormalities after a transient focal neurologic episode may be semantic because, in either case, embolus or atherosclerosis may be responsible for the following stroke and the evaluation for the source of the difficulty is essentially the same in both circumstances.
In the clinical analysis of TIAs, it is also useful to separate a single transient episode from repeated ones that are all of uniform type. The latter are more a warning of impending vascular occlusion, particularly of the internal carotid artery, whereas the former, especially when prolonged, are again often caused by an embolus that leaves no lasting clinical effect. Prolonged, fluctuating TIAs are the most ominous. Approximately 20 percent of infarcts that follow TIAs occur within a month after the first attack, and approximately 50 percent within a year (Whisnant et al). In an attempt to provide a predictive tool, various scales have been devised, among them the “ABCD” system devised by Rothwell and colleagues (2005) and derivatives of this scale. Blood pressure, unilateral weakness, speech disturbance, and the duration of symptoms (all less than 1 h) are added to produce a predictive score for stroke within 1 week. Studies subsequent to the original one have given variable sensitivities, for which reason this interesting approach must be considered in clinical context. In the original study, unilateral weakness and duration lasting over an hour were most predictive of stroke. The problem of determining the cause of a prolonged TIA has been alluded to—many of these cases are a result of emboli.
In a prospective study of a large group of patients with TIAs caused by atherosclerotic vascular disease, the 5-year cumulative rate of fatal or nonfatal cerebral infarction was 23 percent (Heyman et al). Interestingly, the rate of myocardial infarction in this group of patients, particularly in those with carotid lesions, was almost as high (21 percent), and in other series it has exceeded the risk of stroke. Thus the occurrence of carotid TIAs is a predictor not only of cerebral infarction but also of myocardial infarction. About two-thirds of all patients with TIAs are men with hypertension, reflecting the higher incidence of atherosclerosis in this group. Occasionally, in younger adults, TIAs may occur as relatively benign phenomena, without recognizable features of atherosclerosis or risk factors for it. Migraine is suspected in such patients (see further on); other such instances are a result of special hematologic disorders such as the antiphospholipid antibody discussed later in the chapter.
It should also be pointed out that blood diseases that cause excessive viscosity or sludging of blood (polycythemia vera, sickle cell disease, thrombocytosis, leukemia, and hyperglobulinemic states) may also cause TIAs prior to a stroke.
In the transient ischemic attack of the eye, transient monocular blindness (also called amaurosis fugax or TMB) is the usual symptom. Most of the visual episodes evolve swiftly, over 5 to 30 s, and are described as a horizontal shade falling (or rising) smoothly over the visual field until the eye is completely but painlessly blind. The attack clears slowly and uniformly. Sometimes the attack takes the form of a wedge of visual loss, sudden generalized blurring, or, a gray or bright light obscuring vision. Transient attacks of monocular blindness are usually more stereotyped with repeated episodes than are hemispheric attacks. TIAs consisting of a homonymous hemianopia should suggest a stenosis of the posterior cerebral artery but it is often difficult for the patient to make the distinction from monocular blindness.
The implications of amaurosis fugax have been evaluated by several investigators and found not to be quite as ominous as those of hemispheral TIAs, particularly in younger patients. Poole and Ross Russell observed a group of 110 patients for periods of 6 to 19 years following an episode of amaurosis fugax (exclusive of the type caused by cholesterol emboli). At the end of 6 years, the mortality rate (mainly because of heart disease) was 21 percent, but the incidence of stroke was 13 percent (compared to expected figures of 15 and 3, respectively, percent in an age-matched population). Of the patients who were alive at the end of the observation period, 43 percent had had no further attacks of amaurosis fugax following the initial episode. Noteworthy also was the finding that among patients with normal carotid arteriograms, only 1 of 35 had had a stroke during the followup period, whereas stroke had occurred in 8 of 21 patients in whom the internal carotid artery was occluded or stenotic. As pointed out by Benavente and colleagues, the risk of stroke over the 3 years following an attack is as low as 2 percent if there are no other issues such as diabetes, but it may be as high as 24 percent in older patients with risk factors for atherosclerosis. Tippin and coworkers reviewed the records of 83 patients with onset of amaurosis fugax before the age of 45 years and found evidence of stroke in none; moreover, 42 of these patients were examined after a mean period of 5.8 years during which no stroke had occurred. It is evident that in this younger group a mechanism other than atherosclerosis was operative, such as migraine or an antiphospholipid antibody (discussed further on).
It is perhaps not surprising that the risk of stroke after transient monocular blindness is lower than for cerebral TIAs from carotid atherosclerotic disease. The size of particulate material that occludes the ophthalmic and its branches’ vessels is so small that a similar event in the cerebral hemispheres would be less likely to produce symptoms. Furthermore, ischemia of the retina produces symptoms that are hard for the patient to ignore. While there are other underlying causes of TMB, these notions taken together could explain a large part of the difference in risk between conventional TIA and TMB.
It has been recognized that strokes caused by occlusion of small penetrating vessels of the brain have a propensity to be intermittent (“stuttering”) at their onset and occasionally to allow virtually complete restitution of function between discrete episodes. Whether this constitutes a “lacunar TIA” has been debated, but it seems to us that the more important problem is our inability to distinguish a transitory occlusion of a small vessel from that of a larger vessel. Donnan and colleagues (1993) speak of a “capsular warning syndrome,” which we have seen a number of times, consisting of escalating episodes of weakness in the face, arm, and leg and culminating in a capsular lacunar stroke. We conclude that lacunar symptoms at their onset may stutter or remit for hours or days, and there is no doubt that one or many of them may precede a lacunar stroke. Nevertheless, the basic pattern of a small deep stroke remains identifiable in mild form; partial syndromes that simulate cortical TIA are less common. Lacunar stroke is discussed extensively further on.
The question here, so far not satisfactorily answered, is whether reduced blood flow or embolic particles are responsible for TIAs. Whatever the cause of the attacks, they are in most cases intimately related to vascular stenosis and, usually, to ulceration as a result of atherosclerosis and thrombus formation. Embolization of fibrin-platelet material from atherosclerotic sites indeed may be the cause of attacks in some cases, but it is difficult to understand how attacks of identical pattern could be caused by successive emboli from a distance that enter the same arterial branch each time. Moreover, one would expect the involved cerebral tissue to be at least partially damaged by embolism, leaving some residual signs. When a single transient episode has occurred, particularly if prolonged, the factor of recurrence does not enter into the diagnosis, and cerebral embolism must, of course, then be considered. In some cases of documented embolism, the neurologic state fluctuates from normal to abnormal repeatedly for as long as 36 h, giving the appearance of TIAs (“accelerated TIAs”); in others, a deficit of several hours’ duration occurs, fulfilling the traditional (now largely discarded) criterion for TIAs. As already noted, the same sequence of events can precede lacunar infarction and seem far more likely to be the result of locally reduced blood flow than to recurrent emboli. Restated, a single transitory episode, especially if it lasts longer than 1 h, and multiple episodes of different pattern, suggest embolism and must be distinguished from brief (2- to 10-min) recurrent attacks of the same clinical pattern, which suggest TIAs from atherosclerosis and thrombosis in a large vessel.
Ophthalmoscopic observations of the retinal vessels made during episodes of transient monocular blindness may infrequently show either an arrest of blood flow in the retinal arteries and breaking up of the venous columns to form a “boxcar” pattern or scattered bits of white material temporarily blocking the retinal arteries. These observations indicate that in some cases of ischemic attacks involving the retinal vessels, a temporary, complete, or relatively complete cessation of blood flow occurs locally. Whether this is a result of platelet or fibrin emboli or of platelet aggregation in situ because of decreased perfusion pressure remains unsettled.
On the other hand, exercise and postural TIAs, when they do occur, are particularly suggestive of stenosis of aortic branches, as occurs in Takayasu disease (see further on) and in dissection of the aortic arch and, occasionally, in a fixed atherosclerotic carotid stenosis. TIAs induced by hyperventilation are said to be characteristic of moyamoya disease, a progressive stenosis of intracranial vessels discussed in a later section.
In states of anemia, polycythemia, thrombocythemia, extreme hyperlipidemia, hyperviscosity from macroglobulinemia, sickle cell anemia, and extreme hyper- or hypoglycemia, there may be transient neurologic deficits related to rheologic or other changes in blood, as already mentioned. In some of these cases, the metabolic or rheologic change appears to have brought out symptoms of stenosis in a large or small vessel, but just as often the vasculature is normal. Patients with antiphospholipid antibodies may have TIAs, the mechanism of which is undefined.
In some instances the TIAs begin after the artery has already been occluded by thrombus. As shown by Barnett, emboli may arise from the distal end of the thrombus or enter the upper part of the occluded vessel through a collateral artery. However, almost one-fifth of “carotid TIAs” in the series of Pessin and colleagues (1977), and a somewhat larger proportion of cases reported by Ueda and coworkers, had neither stenosis nor ulceration of the carotid arteries. In most of the cases with normal carotid arteries, the ischemic attacks exceeded 1 h in duration, suggesting embolism from the heart or great vessels including the aortic arch; but there were also a small number of brief ischemic attacks that were unexplained even after arteriography.
In general, hemodynamic changes in the retinal or cerebral circulation make their appearance when the lumen of the internal carotid artery is reduced to 2.0 mm or less (normal diameter, 7.0 mm; range, 5 to 10 mm, lower part of this range in women). This corresponds to a reduction in cross-sectional area of the vessel of more than 95 percent. The exact degree of stenosis that may cause TIAs and the risk of stroke with mild and moderate degrees of stenosis are controversial and are addressed further on.
Transient focal neurologic symptoms are ubiquitous in neurologic practice. They may be a result of seizures, migraine, syncope, or other conditions such as transient global amnesia (see Chap. 21), and they occur occasionally in patients with multiple sclerosis. The clinical setting in which they occur assists in making clear the nature of the attack. Furthermore, transient and reversible episodes of focal cerebral symptoms, indistinguishable from TIAs, are known to occur in patients with meningioma, glioblastoma, metastatic brain tumors situated in or near the cortex, and even with subdural hematoma. Although infrequent, these attacks are important mainly because the use of anticoagulants is relatively contraindicated in some of these circumstances. We have seen these episodes mainly with meningiomas and subdural hematomas; they have consisted of transient aphasia or speech arrest lasting from 2 min to several hours, but sensory symptoms with or without spread over the body, arm weakness, and hemiparesis have also been reported. Some remarkable cases of meningioma have involved repeated transient attacks for decades. Seizures are always suspected in these cases but are rarely proved. It has been speculated that a local vascular disturbance of some kind is operative, but the mechanism is not understood. As far as we can determine, mass lesions have not caused episodes that simulate posterior circulation TIAs.
The issue of the uncertainty regarding vertigo alone as a manifestation of a TIA referable to the basilar or vertebral artery was addressed in Chap. 15. There are occasional cases in which multiple brief episodes of vertigo, lasting perhaps a minute or less and fluctuating in intensity, may be interspersed with additional signs of brainstem ischemia. Careful questioning of the patient usually settles the question but imaging may be necessary in cases where uncertainty remains. Even then, more instances of vertigo than are justified are attributed to atherosclerotic disease in the posterior vessels. In some patients, the complaint of “dizziness” will prove, however infrequently, to be part of a carotid TIA; hence this symptom, in our experience and that of Ueda and associates, is not a totally reliable indicator of the vascular territory involved. According to Ross Russell, so-called drop attacks (see Chap. 7) have been recorded in 10 to 15 percent of patients with vertebrobasilar insufficiency but we have never observed such attacks as a recurrent ischemic phenomenon or a manifestation of other forms of cerebrovascular disease and the syndrome has usually been due to syncope, seizure, or has been of obscure origin.
Cerebral infarction basically comprises two pathophysiologic processes: one, a loss of the supply of oxygen and glucose secondary to vascular occlusion, and the other, an array of changes in cellular metabolism consequent to the collapse of energy-producing processes, ultimately with disintegration of cell structures and their membranes, a process subsumed under the term necrosis. Of potential therapeutic importance are the observations that some of the cellular processes leading to neuronal death are not irrevocable and may be reversed by early intervention, either through restoration of blood flow, by prevention of the influx of calcium into cells, or by interdicting intermediary processes involved in cell death.
At the center of an ischemic stroke is a zone of infarction. The necrotic tissue swells rapidly, mainly because of excessive intracellular water content (cytotoxic edema). Because anoxia also causes necrosis and swelling of cerebral tissue, oxygen lack must be a factor common to both infarction and anoxic encephalopathy. The effects of ischemia, whether functional and reversible or structural and irreversible, depend on its degree and duration. The margins of the infarct are hyperemic, being supplied by meningeal collaterals, and here there is only minimal or no parenchymal damage.
Implicit in discussions of ischemic stroke and its treatment is the existence of a “penumbra” zone that is marginally perfused and contains at-risk but viable neurons. Presumably this zone exists at the margins of an infarction, which at its core has irrevocably damaged tissue that is destined to become necrotic. Using various methods, such a penumbra can be demonstrated in association with some infarctions but not all, and the degree of reversible tissue damage is difficult to determine. The neurons in the penumbra are considered to be physiologically “stunned” by moderate ischemia and subject to salvage if blood flow is restored in a certain period of time. Olsen and colleagues demonstrated hypoperfused penumbral zones but, interestingly, found that regions just adjacent to them are hyperperfused. These concepts find a parallel in attempts to demonstrate by imaging matching of perfusion and infarction (detected by diffusion-weighted images on MRI) in patterns with acute stroke as discussed in the section on treatment. Elevating the systemic blood pressure or improving the rheologic flow properties of blood in small vessels by hemodilution improves flow in the penumbra; however, attempts to use these techniques in clinical work have met with mixed success.
The effects of a focal arterial occlusion on brain tissue vary depending on the location of the occlusion and on available collateral and anastomotic channels. In occlusion of the internal carotid artery in the neck, there may be anastomotic flow through the anterior and posterior communicating arteries of the circle of Willis from the external carotid artery through the ophthalmic artery or via other smaller external-internal connections (Fig. 34-1). With blockage of the vertebral artery, the anastomotic flow may be via the deep cervical, thyrocervical, or occipital arteries or retrograde from the other vertebral artery and again through the posterior communicating arteries. If the occlusion is in the stem portion of one of the cerebral arteries, i.e., distal to the circle of Willis, a series of meningeal interarterial anastomoses may carry sufficient blood into the compromised territory to lessen ischemic damage (Fig. 34-2). There is also a capillary anastomotic system between adjacent arterial branches, and although it may reduce the size of an ischemic territory, it is usually not adequate to prevent infarction. Thus, in the event of occlusion of a major arterial trunk, the extent of infarction ranges from none at all to the entire vascular territory of that vessel. Between these two extremes are all degrees of variation in the extent of infarction and its degree of completeness.
Figure 34-1.
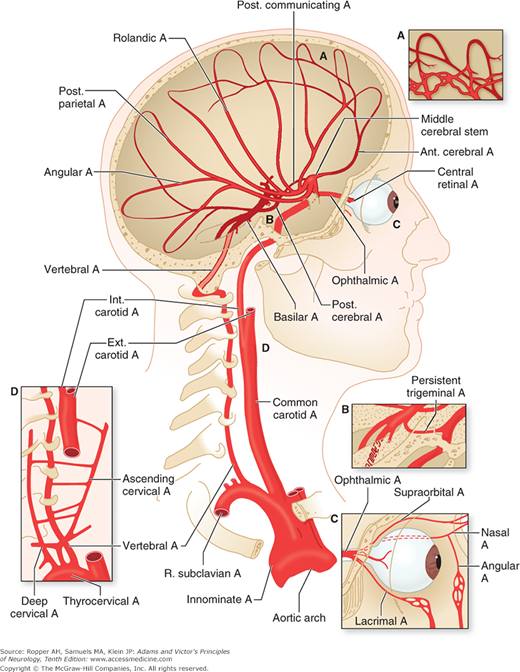
Arrangement of the major arteries on the right side carrying blood from the heart to the brain. Also shown are collateral vessels that may modify the effects of cerebral ischemia. For example, the posterior communicating artery connects the internal carotid and the posterior cerebral arteries and may provide anastomosis between the carotid and basilar systems. Over the convexity, the subarachnoid interarterial anastomoses linking the middle, anterior, and posterior cerebral arteries are shown, with inset A illustrating that these anastomoses are a continuous network of tiny arteries forming a border zone between the major cerebral arterial territories. Occasionally a persistent trigeminal artery connects the internal carotid and basilar arteries proximal to the circle of Willis, as shown in inset B. Anastomoses between the internal and external carotid arteries via the orbit are illustrated in inset C. Wholly extracranial anastomoses from muscular branches of the cervical arteries to vertebral and external carotid arteries are indicated in inset D.
Figure 34-2.
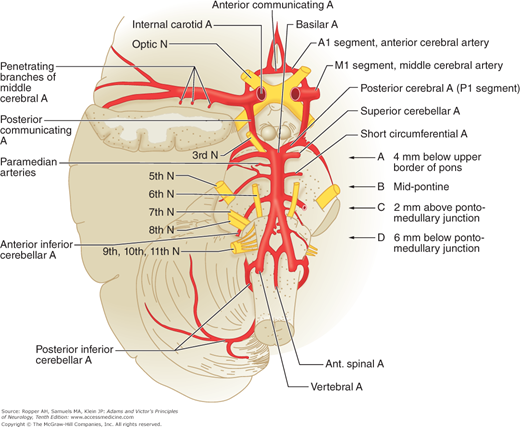
Diagram of the base of the brain showing the principal vessels of the vertebrobasilar system (the circle of Willis and its main branches). The term M1 is used to refer to the initial (stem) segment of the middle cerebral artery; A1 to the initial segment of the anterior cerebral artery proximal to the anterior communicating artery; A2 to the postcommunal segment of the anterior cerebral artery; and P1 and P2 to the corresponding pre- and post-communicating segments of the posterior cerebral artery. The letters and arrows on the right indicate the levels of the four cross-sections following: A = Fig. 34-15; B = Fig. 34-14; C = Fig. 34-13; D = Fig. 34-12. Although vascular syndromes of the pons and medulla have been designated by sharply outlined shaded areas, one must appreciate that because satisfactory clinicopathologic studies are scarce, the diagrams do not always represent established fact. The frequency with which infarcts fail to produce a well-recognized syndrome and the special tendency for syndromes to merge with one another must be emphasized.
The phenomenon of cerebrovascular autoregulation is appropriately introduced here. Over a range of mean blood pressures of approximately 50 to 150 mm Hg, the small pial vessels are able to dilate and to constrict in order to maintain cerebral blood flow (CBF) in a relatively narrow range. This accommodation eventually fails at the extremes of blood pressure, after which CBF follows systemic pressure passively, either falling precipitously or rising to levels that damage the walls of small vessels. The conditions in which the limits of autoregulation are exceeded are at the extremes of hypertensive encephalopathy at one end and circulatory failure at the other, both of which are discussed in later sections of the chapter.
If brain tissue is observed in experimental circumstances at the time of arterial occlusion, the venous blood is first seen to darken, owing to an increase in deoxygenated hemoglobin. The viscosity of the blood and resistance to flow both increase, and there is sludging of formed blood elements within vessels. The tissue becomes pale. Arteries and arterioles become narrowed. Upon reestablishing flow in the occluded artery, the sequence is reversed and there may be a slight hyperemia.
Many of these factors relating to cerebral blood flow have been studied by Heiss and by Siesjo and others and are reviewed in detail by Hossman. They have determined the critical threshold of CBF, measured by xenon clearance, below which functional impairment occurs. In several animal species, including macaque monkeys and gerbils, the critical level was 23 mL/100 g/min (normal is 55); if, after a short period of time, CBF is restored to higher levels, the impairment of function can be reversed. Reduction of CBF below 10 to 12 mL/100 g/min causes infarction, almost regardless of its duration. The critical level of hypoperfusion that abolishes function and leads to tissue damage is therefore a CBF between 12 and 23 mL/100 g/min. At these levels of blood flow the electroencephalogram (EEG) is slowed, and below this level it becomes isoelectric. In the region of marginal perfusion, the K level increases (as a result of efflux from injured depolarized cells) and adenosine triphosphate (ATP) and creatine phosphate are depleted. These biochemical abnormalities are reversible if the circulation is quickly restored to normal. Disturbance of calcium ion homeostasis and accumulation of free fatty acids interfere with full recovery of cells. A CBF of 6 to 8 mL/100 g/min causes marked ATP depletion, increase in extracellular K, increase in intracellular Ca, and cellular acidosis, invariably leading to histologic signs of necrosis. These changes do not become apparent for several hours. Free fatty acids (appearing as phospholipases) are activated and destroy the phospholipids of neuronal membranes. Prostaglandins, leukotrienes, and free radicals accumulate, and intracellular proteins and enzymes are denatured. Cells then swell, a process called cellular, or cytotoxic, edema (see “Brain Edema” in Chap. 31). Similar abnormalities affect mitochondria even before other cellular changes are evident.
Regarding anoxic damage of the brain, Ames and Nesbett, in a series of articles, studied the rabbit retina in an immersion chamber in which O2 and various substrates could be altered directly rather than through the vasculature. They found that cells could withstand complete absence of O2 for 20 min. After 30 min of anoxia, there was irreversible damage, reflected by an inability of the tissue to utilize glucose and to synthesize protein. Hypoglycemia further reduced the tolerance to hypoxia, whereas the tolerance could be prolonged by reducing the energy requirements of cells (increasing magnesium in the medium). Ames and colleagues (1968) postulated that the long period of tolerance of retinal neurons to complete anoxia in vitro, in comparison to that in vivo, is related to what he called the no-reflow phenomenon (swelling of capillary endothelial cells, which prevents the restoration of circulation), as mentioned earlier. Body temperature is yet another important factor in determining the extent of infarction. A reduction of even 2 to 3°C (3.6 to 5.4°F) reduces the metabolic requirements of neurons and increases their tolerance to hypoxia by 25 to 30 percent.
One area of interest has focused on the role of excitatory neurotransmitters in stroke, particularly glutamate and aspartate, which are formed from glycolytic intermediates of the Krebs cycle. These neurotransmitters, released by ischemic cells, excite neurons and produce an intracellular influx of Na and Ca. These changes are in part responsible for irreversible cell injury, but this must be an oversimplification. Some current attempts at therapy, for example, are directed at limiting the extent of infarction by blocking the glutamate receptor, particularly the NMDA (N-methyl-D-aspartate) channel—one of several calcium channels that open under conditions of ischemia and set in motion a cascade of cellular events eventuating in neuronal death (apoptosis). However, even complete blockade of the NMDA channels has not prevented cellular death, presumably because dysfunction of several other types of calcium channels continues and allows calcium entry to cells. Additional biochemical events must be induced by ischemia, including the production of free radicals, which leads to peroxidation and disruption of the outer cell and mitochondrial membranes. Clearly, the cascade of intracellular events that lead to neuronal death is likely to be more complex than is currently envisioned.
The extent of neural tissue dysfunction is not dictated solely by the activation of these mechanisms in neurons. It is now clear that toxic influences are exerted on oligodendroglial cells in white matter during ischemia and on astrocytic cells that support neurons. Moreover, injury to both neurons and astroyctes is augmented by an inflammatory response that activates endothelial cells to express cell adhesion molecules that attract additional inflammatory cells and upregulate levels of inflammatory proteases (e.g., metalloproteases) and cytokines (e.g., interleukins and chemokines). These events are summarized in the review by Lo and coworkers and are areas of active research.
It is also clear that as ATP production fails, there is significant accumulation of lactic acid in cerebral tissue, and all the biochemical changes consequent to the cellular acidosis occur. These may also be of importance in determining the extent of cell damage (see reviews of Raichle and of Plum). Myers and Yamaguchi showed that monkeys infused with glucose before the induction of cardiac arrest suffered more brain damage than did either fasted or saline-infused animals. They suggested that the high cerebral glucose level under anaerobic conditions led to increased glycolysis during the ischemic episode and that the accumulated lactate was neurotoxic. On the basis of such observations, Plum suggested that scrupulous control of the blood glucose might reduce the risk of cerebral infarction in diabetic and other stroke-prone patients, and during conditions of potential hyperglycemia. Clinical implementation of this idea is difficult and its advantages remain to be established. Nonetheless, these multiple molecular pathways for neuronal damage provide opportune points for therapeutic intervention.
Involved in the process of thrombosis are changes in a number of natural anticoagulant factors such as heparin cofactor 2, antithrombin III, protein C, and protein S. Some of these are extrinsic to the blood vessels and hence may result in thrombosis in one or in multiple sites even without prior vascular injury. These are discussed by Furie and Furie. Protein C is a vitamin K-dependent protease that, in combination with its cofactors protein S and antithrombin III, inhibits coagulation. A deficiency of any of these factors may predispose to in situ thrombosis within either the arterial or venous systems and is a cause of otherwise unexplained strokes in young persons. For example, protein C deficiency (heterozygous in one of every 16,000 individuals) is a cause of thrombosis of both veins and arteries; a resistance to activated protein C has also been described (causing venous thrombosis almost exclusively). Antiphospholipid antibody is yet another cause of vascular occlusion that is not incited by damage to the vessel wall (see later in chapter). The metabolic disturbances in a number of metabolic diseases such as Fabry disease also favor cerebrovascular clotting. Persons with inflammatory bowel diseases (ulcerative colitis, Crohn disease) are known to be prone to thrombotic strokes. Whether inflammation elsewhere in the body predisposes to cerebral vascular occlusions is an open question. Curiously, the hypercoagulable state induced by certain adenocarcinomas (Trousseau’s syndrome) does not often produce in situ arterial occlusion but it does lead to thrombotic vegetations on heart valves that precipitate strokes and it predisposes to cerebral venous thrombosis as discussed further on.
These hematologic factors should be sought when unexplained strokes occur in children or young adults, in families whose members have had frequent strokes, in pregnant or parturient women, and in women who are migraineurs or taking birth control pills. According to Markus and Hambley, whose review of this subject is recommended, screening for lupus anticoagulant, anticardiolipin antibodies, deficiency of proteins C and S, and antithrombin III is probably justified, but mainly in these special circumstances. This special category of vascular thrombosis is taken up in later sections.
Imaging Techniques in Stroke
Technologic advances continue to enhance the clinical study of stroke patients; they allow the demonstration of both the cerebral lesion and the affected blood vessel. CT demonstrates and accurately localizes even small hemorrhages, hemorrhagic infarcts, subarachnoid blood, clots in and around aneurysms, arteriovenous malformations, and established regions of infarction.
Magnetic resonance imaging is able to reveal flow voids in vessels, hemosiderin and iron pigment, and the alterations resulting from ischemic necrosis and gliosis. MRI is particularly advantageous in demonstrating small lacunar lesions deep in the hemispheres and abnormalities in the brainstem (a region obscured by adjacent bone in CT). However, the main advance has been the introduction of diffusion-weighted magnetic resonance techniques, which allow the detection of an infarctive lesion within minutes of the stroke, i.e., considerably earlier than CT and other MRI sequences (Fig. 34-3). The various MRI imaging sequences used in the diagnosis and dating of stroke are discussed below and in Chap. 2 and in Table 34-4.
Figure 34-3.
MRI showing acute infarctions. The upper images show a right middle cerebral artery infarction that appears bright on diffusion-weighted imaging (DWI) (upper left). There is subtle hyperintensity representing early vasogenic edema on T2-FLAIR sequence (upper right). The lower images show an acute cerebellar infarction in the territory of the posterior inferior cerebellar artery (PICA) that is bright on DWI (lower left) and faintly bright on T2-FLAIR (arrow, lower right). There is also a previous infarction just anterior to the acute cerebellar stroke that is dark on DWI and bright on T2 due to gliosis.
SEQUENCE | TE | TR | GOOD FOR | BRIGHT | DARK |
|---|---|---|---|---|---|
T1-weighted | <50 | <1,000 | Subacute blood, contusions, CSF disorders | CSF, bone, edema, deoxyhemoglobin mineralization | |
T2-weighted | >80 | >2,000 | Infarcts, inflammation, tumors | CSF, liquids, edema | Solids, calcium |
Proton (spin) density | <50 | >2,000 | “Most pathology” | Fat, water, acute blood | |
Fluid-attenuated inversion recovery (FLAIR) | >80 | >10,000 | Edema, inflammation | Edema, inflammation, gliosis | CSF; otherwise like T2 |
Diffusion-weighted image (DWI) | Acute infarcts | Blood after several days | |||
Susceptibility sequence | Hemorrhages, calcification | Blood | |||
Short tau inversion recovery (STIR) | Spine and orbit studies (eliminates fat signals) |
Arteriography, enhanced by digital processing of images, accurately demonstrates stenoses and occlusions of the intracranial and extracranial vessels as well as aneurysms, vascular malformations, and other blood vessel diseases such as arteritis and vasospasm. To a large extent, conventional contrast angiography has been supplanted by magnetic resonance angiography (MRA), venography (MRV), and CT angiography for the visualization of large intracranial arteries and veins (see Fig. 2-4). These techniques have the advantage of relative safety (injection of contrast media is required for CT angiography) but do not give refined images of the smaller blood vessels. MRA depicts the “time of flight” of blood through vessels and is not as accurate as CT angiography in measuring the degree and morphology of changes within a cerebral or intracranial vessel. Vascular imaging by MRI has the advantage that areas of restricted diffusion can be detected during the same imaging session, while the CT technique, in addition to better resolution of vascular lumens, is preferable in circumstances requiring the demonstration of soft tissues and bone adjacent to blood vessels, thereby providing the surgeon with important anatomic information (see Chap. 2).
Other procedures for the investigation of cerebrovascular disease include Doppler ultrasound flow studies, which demonstrate atheromatous plaques and stenoses of large vessels, particularly of the carotid but also of the vertebrobasilar arteries. The transcranial Doppler technique has reached a degree of precision whereby occlusion or spasm of the main vessels of the circle of Willis can be detected and roughly quantitated. Various methods of measuring regional blood flow with positron emission tomography (PET) and radionuclide imaging find use in special instances discussed in appropriate sections of the chapter.
The EEG and lumbar puncture have lost favor in stroke diagnosis but the former is possibly underused as a readily available means of detecting cortical infarction in the wake of ischemia of a region of one hemisphere. It allows a distinction to be made between occlusion of a small and a large vessel, because focal EEG abnormalities are sparse or absent with a deep lacunar stroke. The technique finds more use in distinguishing between transient alterations in nervous function that are a result of seizures and episodes caused by focal ischemia.
Whereas most cerebral arteries can be evaluated only indirectly, more direct clinical means of physical examination are available for the evaluation of the common and internal carotid arteries in the neck. With severe atherosclerotic stenosis at the level of the carotid sinus auscultation discloses a bruit, best heard with the bell of the stethoscope held against the skin just tightly enough to create a seal (excessive pressure creates a diaphragm of the skin and filters the low-pitched frequencies that are typical of the bruit of carotid stenosis). Occasionally, a bruit at the angle of the jaw is caused by stenosis at the origin of the external carotid artery or is a radiated murmur from the aortic valve and can then be misleading. If the bruit is loudest at the angle of the jaw, the stenosis usually lies at the proximal internal carotid; if heard lower in the neck, it is in the common carotid or subclavian artery and may be radiated from the aortic valve. Rarely, stenosis in vertebral arteries or vascular malformations at the base of the brain produce bruits that are heard posteriorly in the neck.
The presence of a bruit in the neck is an indication of cerebrovascular disease but its detection is not highly correlated with the presence of severely stenotic lesions as assessed by ultrasonography or angiography. In the past, the physician was almost completely dependent on the details of the nature of the bruit but these details are now of limited interest. An additional though infrequent sign of carotid occlusion is the presence of a bruit over the opposite carotid artery, heard by placing the bell of the stethoscope over the eyeball (ocular bruit). As pointed out by Pessin and colleagues (1983), this murmur is often caused by augmented circulation through the patent vessel but there have been as many instances in our experience when a bruit over the eye instead reflects a stenosis in the intracranial portion of the carotid artery on that side. These and other tests for assessing carotid flow have been supplanted by ultrasound insonation and imaging of the carotid artery, but retinal examination remains valuable in that it may demonstrate emboli within retinal arteries, either shiny white or reddish in appearance; this is another important sign of carotid disease (crystalline cholesterol, termed Hollenhorst plague, is sloughed from an atheromatous ulcer).
The Stroke Syndromes
The clinical picture that results from an occlusion of any one artery differs in minor ways from one patient to another, but there is sufficient uniformity to justify the assignment of a typical syndrome to each of the major cerebral arteries and their branches and their identification by careful examination of highly specific neurovascular syndromes is one of the cardinal skills of the clinical neurologist. The following descriptions apply particularly to the clinical effects of ischemia and infarction caused by embolism and thrombosis. The distinction between vascular occlusion from a local atherosclerotic plaque with superimposed thrombosis and an embolic occlusion is made largely on the basis of factors already enumerated: (1) the temporal profile of the stroke syndrome, an immediate stroke favoring embolus, and a slowly evolving or “stuttering” onset or emergence form sleep with a stroke favoring atherosclerosis, and (2) associated medical risk factors such as atrial fibrillation (strongly favoring embolus) or diabetes, hypertension, hyperlipidemia and smoking, together favoring atherosclerosis of the small penetrating, or large trunk vessels. Although hemorrhage within a specific vascular territory may give rise to many of the same effects, the total clinical picture is different because it usually involves regions supplied by more than one artery and, by its deep extension and pressure effects, causes secondary features of headache, vomiting, and hypertension, as well as a series of falsely localizing signs, as described in Chaps. 17 and 31.
The carotid system consists of three major arteries: the common carotid, internal carotid, and external carotid. As indicated in Fig. 34-1, the right common carotid artery arises at the level of the sternoclavicular notch from the innominate (brachiocephalic) artery, and the left common carotid comes directly from the aortic arch. The common carotid arteries ascend in the neck to the C4 level, just below the angle of the jaw, where each divides into external and internal branches (sometimes the bifurcation is slightly above or below this point). This part of the extracerebral circulation is essential to an understanding of stroke. The carotid vessels are subject to atherosclerotic narrowing, atherothrombotic occlusion, arterial dissection and rarely, other processes such as various forms of vasculitis.
Occlusion of the common carotid artery accounts for less than 1 percent of cases of carotid artery syndrome, the remainder being because of disease of the internal carotid artery itself. Nevertheless, the common carotid can be occluded by an atheromatous plaque at its origin in the thorax, more often on the left side. Atherosclerotic stenosis or occlusion of the midportion of the common carotid may also occur years after radiation therapy for laryngeal, thyroid, or other head and neck cancer. If the bifurcation is patent, few if any symptoms may result because retrograde flow from the external carotid maintains internal carotid flow and perfusion of the brain.
The remainder of this discussion is concerned with disease of the internal carotid artery. The territory supplied by this vessel and its main branches is shown in Figs. 34-1, 34-4 and 34-5.
Figure 34-4.
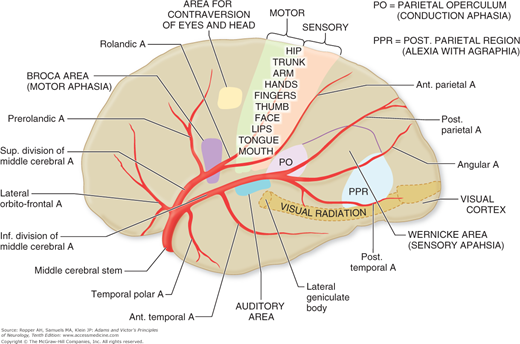
Diagram of the left cerebral hemisphere, lateral aspect, showing the courses of the middle cerebral artery and its branches and the principal regions of cerebral localization. Below is a list of the clinical manifestations of infarction in the territory of this artery and the corresponding regions of cerebral damage.
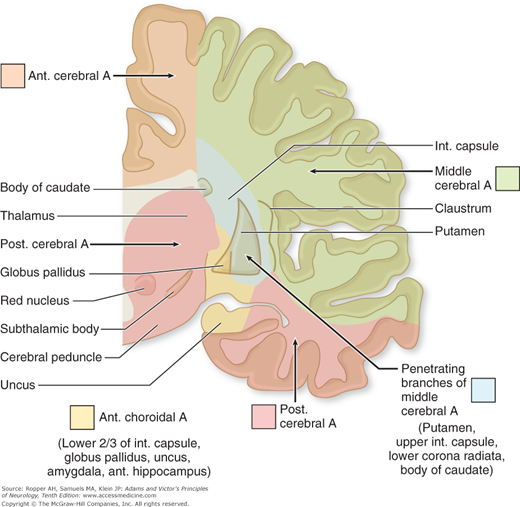
The territory affected by diminished blood flow in the brain in cases of carotid occlusion is highly dependent on the configuration of the circle of Willis. For example, when the anterior communicating artery is very small, the ipsilateral anterior cerebral territory is affected as well. In extreme instances where the circle of Willis provides no communication to the side of an occluded carotid artery, thus isolating the hemisphere from other blood flow, massive infarction involving the anterior two-thirds or all of the cerebral hemisphere results. If the two anterior cerebral arteries arise from a common stem on one side, infarction may occur in the territories of both vessels. The territory supplied by the posterior cerebral artery will also be included if this vessel is supplied by the internal carotid rather than the basilar artery (a configuration that reflects a residual fetal origin of the posterior cerebral artery). The clinical manifestations of atherosclerotic thrombotic disease of this artery are among the most variable of any cerebrovascular syndrome because the internal carotid is not an end vessel. In most individuals it is in continuity with the vessels of the circle of Willis and those of the orbit, and no part of the brain is completely dependent on it. Therefore occlusion, which occurs most frequently in the first part of the internal carotid artery immediately beyond the carotid bifurcation, may be silent (30 to 40 percent of cases).
If one internal carotid artery had been occluded at an earlier time, occlusion of the other may cause bilateral cerebral infarction. The clinical effects in such cases may include coma with quadriplegia and continuous horizontal “metronomic” conjugate eye movements.
Occlusion of the distal intracranial portion of the internal carotid artery (the “T”)—for example by an embolus to its distal part—produces a clinical picture like that of middle cerebral artery occlusion: contralateral hemiplegia, hemihypesthesia, and aphasia (with involvement of the dominant hemisphere). When the anterior cerebral territory is included, there are additional clinical features of leg paralysis as described further on. Patients with such large infarctions are usually immediately drowsy or stuporous because of an ill-defined effect on the reticular activating system.
Headache, located as a rule above the eyebrow, on the side of the infarction, may occur with thrombosis or embolism of the carotid artery, but cranial pain is not invariable and is usually mild. The headache associated with occlusion of the middle cerebral artery tends to be more lateral, at the temple; that of posterior cerebral occlusion is located in or behind the eye.
When the circulation of one carotid artery has been incompletely compromised, reducing blood flow in both the middle and anterior cerebral territories on that side, the zone of maximal ischemia lies between the two vascular territories (“cortical watershed”) or, alternatively, in the deep portions of the hemisphere between the territories of the lenticulostriate branches and the penetrating vessels from the convexity (“internal” or “deep watershed”). The infarction in the first instance occupies a region in the high parietal and frontal cortex and the adjacent subcortical white matter. Its size depends upon the adequacy of collateral vessels. Weakness tends to involve the shoulder and hip more than the hand and face. With long-standing carotid stenosis, the cortical watershed zone shifts downward toward the perisylvian portions of the middle cerebral artery territory, even to the extent that a stroke may weaken facial movement or cause a nonfluent aphasia. With impaired perfusion of the deep watershed, infarctions of varying size are situated in the subfrontal and subparietal portions of the centrum semiovale.
The situation is somewhat different in cases of total circulatory collapse from cardiac arrest, in which perfusion fails not only in the watershed areas between the middle and anterior cerebral arteries but also between the middle and posterior cerebral arteries. Bilateral infarctions are then situated within a zone that extends in a sickle-shaped strip of variable width from the cortical convexity of the frontal lobe through the high parietal lobe, to the occipitoparietal junction. Deeper infarctions also occur, but they more often take the form of contiguous extensions of the just described cortical infarction into the subjacent white matter. There may appear to be several separate infarctions after hypoperfusion states, but these often turn out to be radiographically visible portions of a larger border-zone lesion.
The internal carotid artery also nourishes the optic nerve and retina. For this reason, transient monocular blindness occurs prior to the onset of stroke in 10 to 25 percent of cases of symptomatic carotid occlusion. Yet central retinal artery ischemia is a relatively rare manifestation of carotid artery occlusion, presumably because of efficient collateral supply in the globe.
Signs of carotid occlusion include transient monocular blindness or visual loss or dimness of vision with exercise, after exposure to bright light, or on assuming an upright position; retinal atrophy and pigmentation; atrophy of the iris; peripapillary arteriovenous anastomoses in the retinae; and claudication of jaw muscles. However, the cardinal clinical signs of stenoses, ulcerations, and dissections of the internal carotid artery are TIAs. It is a subject of debate whether these are the result of fibrin platelet emboli or a reduction in blood flow. TIAs were discussed earlier, but here it can be stated again that they represent an important risk factor for stroke.
The middle cerebral artery (MCA) has superficial and deep hemispheral branches that together supply the largest portion of the cerebral hemisphere. Through its cortical branches, it supplies the lateral (convexity) part of the cerebral hemisphere (see Fig. 34-4) encompassing (1) the cortex and white matter of the lateral and inferior parts of the frontal lobe—including motor areas 4 and 6, contraversive centers for lateral gaze and the motor speech area of Broca (dominant hemisphere); (2) the cortex and white matter of the parietal lobe, including the primary and secondary sensory cortices and the angular and supramarginal gyri; and (3) the superior parts of the temporal lobe and insula, including the receptive language area of Wernicke. The deep penetrating or lenticulostriate branches of the MCA supply the putamen, a large part of the head and body of the caudate nucleus (shared with the Heubner artery, see further on), the outer globus pallidus, the posterior limb of the internal capsule, and the corona radiata (see Fig. 34-5).
The MCA may be occluded in its longitudinal portion, or the stem, that is proximal to its bifurcation (the term M1 is used by radiologists to denote this portion of the vessel). An occlusion at this site blocks the flow in the small deep penetrating vessels as well as in superficial cortical branches. An occlusion at the distal end of the stem blocks only the orifices of the divisions of the artery in the sylvian sulcus but leaves unaffected the deep penetrating vessels. The picture of total occlusion of the stem is one of contralateral hemiplegia (involving the face, arm, and leg as a result of infarction of the posterior limb of the internal capsule), hemianesthesia, and homonymous hemianopia (because of infarction of the lateral geniculate body), with deviation of the head and eyes toward the side of the lesion. In addition, there is a variable but usually global aphasia with left hemispheric lesions and anosognosia and amorphosynthesis with right-sided lesions (see Chap. 22). In the beginning, the patient may be drowsy or stuporous because of an ill-defined effect of widespread paralysis of neurologic function. Once fully established, the motor, sensory, and language deficits remain static or improve little as months and years pass. If the patient is globally aphasic for many months, he seldom ever again communicates effectively (see Chap. 23 for a discussion of the aphasic disorders). If there are adequate collateral vessels over the surface of the hemisphere, only those components of the stroke referable to the deep structures are evident (mainly hemiplegia encompassing the contralateral limbs and face) as discussed below, the cortical elements of aphasia, agnosia, and apraxia then being absent or mild.
Occlusion of the stem of the MCA is usually caused by embolus and less often by a thrombus superimposed on an atherosclerotic plaque. Studies over the years have affirmed that most carotid occlusions are thrombotic, whereas most middle cerebral occlusions are embolic (Fisher, 1975; Caplan, 1989). The emboli may lodge in the stem or, more often, drift into the cortical branches as described below; not more than 1 in 20 will enter deep penetrating branches that originate in the stem. In the less-common circumstance, the MCA may become stenotic from atherosclerosis. The stroke is then the result of occlusion of the vessel by a superimposed thrombus. In several series of such cases, the stroke was preceded by TIAs, producing a picture resembling that of carotid stenosis (see Caplan, 1989). Transient monocular blindness does not occur in this situation because the occlusion is distal to the ophthalmic artery. In epidemiologic studies, certain populations such as Asians are disproportionally affected by this form of intracranial atherosclerosis, as are diabetics.
A number of interesting syndromes occur with deep lesions in the territory of the penetrating, lenticulostriate, vessels of the MCA (see Figs. 34-5 and 34-7). Most, as mentioned, are attributable to emboli that lodge in the stem of the main vessel, although imaging studies may show a patent vessel and others are undoubtedly atherothrombotic. There have been few adequate pathologic studies. Although the infarction is centered in the deep white matter, most of the syndromes are fragments of the cortical–subcortical stroke patterns described further on. The most common type in our experience has been a large striatocapsular infarction, similar to that described by Weiller and colleagues. All of their patients had a degree of hemiparesis and one-fifth had aphasia or hemineglect. Aphasia, when it occurred, tended to be a limited form of the Broca type and in our experience, has been short-lived. With smaller deep strokes we have most often encountered incomplete motor syndromes affecting only the arm and hand, without language disturbance or neglect; these are difficult to differentiate from small embolic cortical strokes. The lesions in the corona radiata are larger than typical lacunar infarctions (see further on) but probably have a similar pathophysiology.
Homonymous hemianopia may occur with posterior capsular lesions, probably as a result of damage to the region of the lateral geniculate nucleus, but it is infrequent and must be distinguished from visual hemineglect of contralateral space. Bilateral cerebral infarctions involving mainly the insular–perisylvian (anterior opercular) regions manifest themselves by a diplegia of the face, tongue, and masseters that results in anarthria without aphasia (see Mao et al).
An embolus entering the middle cerebral artery most often lodges in one of its two main branches, the superior division (supplying the rolandic and prerolandic areas) or the inferior division (supplying the lateral temporal and inferior parietal lobes). Atherothrombotic occlusion of this vessel is infrequent. Major infarction in the territory of the superior division causes a dense sensorimotor deficit in the contralateral face, arm, but, to a lesser extent the leg, as well as ipsilateral deviation of the head and eyes; i.e., it differs from the MCA stem occlusion syndrome in that the leg and foot are partly spared and less involved with weakness than the arm and face (“brachiofacial,” or chierobrachial “paralysis”); there is no impairment of alertness. If the occlusion is long-lasting (not merely transient ischemia with disintegration of the embolus) there will be slow improvement; after a few months, the patient is able to walk with a spastic leg, while the motor deficits of the arm and face remain. The sensory deficit may be profound, resembling that of a thalamic infarct (as described in Chaps. 8 and 9) but more often it is less severe than the motor deficit, taking the form of stereoanesthesia, agraphesthesia, impaired position sense, tactile localization, and two-point discrimination, as well as variable changes in touch, pain, and temperature sense (see Chap. 22). With left-sided lesions there is initially a global aphasia, which changes to a predominantly nonfluent (Broca’s) aphasia, with the emergence of an effortful, hesitant, grammatically simplified, and dysmelodic speech (see Chap. 23); or quite often there is Broca’s aphasia from the outset.
Embolic occlusion limited to one of the distal branches of the superior division, perhaps the most common stroke seen in clinical practice, produces a more circumscribed infarct that further fractionates the above-described syndrome. With occlusion of the ascending frontal branch, the motor deficit is limited to the face and arm with little or no weakness of the leg, and the latter, if weakened at all, soon improves; with left-sided lesions, there is dysfluent and agrammatic speech and normal comprehension (Broca’s aphasia). Embolic occlusion of the left rolandic branch alone results in sensorimotor paresis with severe dysarthria but little evidence of aphasia. A cortical–subcortical branch occlusion may give rise solely to a brachial monoplegia or hand weakness that simulates a problem in the peripheral nervous system. Embolic occlusion of ascending parietal and other posterior branches of the superior division may cause no sensorimotor deficit but only a conduction aphasia (see Chap. 23) and ideomotor apraxia. There are many other limited stroke syndromes or combinations of the aforementioned deficits relating to small regions of damage in the frontal, parietal, or temporal lobes. Among these are the Gerstmann syndrome and various forms of agnosia (in some patients, these may be in the territory of the inferior division of the MCA discussed below). Most of these are discussed in Chap. 22, which details the result of lesions in particular parts of the cerebrum. Improvement can be expected within a few weeks to months but some remnant of the original problem usually remains in place. As indicated earlier, the distal territory of the middle cerebral artery may also be rendered ischemic by failure of the systemic circulation, especially if the carotid artery is stenotic; this situation may simulate embolic branch occlusions.
Occlusion of the inferior division of the MCA is slightly less frequent than occlusion of the superior one, but again is nearly always the result of embolism. The usual result in left-sided lesions is a Wernicke’s aphasia, which generally remains static for days or weeks after which some improvement can be expected. In less-extensive infarcts that are the result of selective distal branch occlusions (superior parietal, angular, or posterior temporal), the deficit in comprehension of spoken and written language may be especially severe. Again, after a few months, the deficits usually improve, often to the point where they are evident only in self-generated efforts to read and copy visually presented words or phrases. With either right- or left-hemispheric lesions, there is usually a superior quadrantanopia or homonymous hemianopia and, with right-sided ones, a left visual neglect and other signs of amorphosynthesis. Rarely, an agitated confusional state, presumably from nondominant temporal lobe damage, may be a prominent feature of dominant hemispheral lesions and sometimes of nondominant ones. Some of the syndromes applicable to the angular gyrus and the supramarginal gyrus may occur in strokes within this division, depending on the distributions of the vessels in an individual.
This artery, through its cortical branches, supplies the anterior three-quarters of the medial surface of the frontal lobe, including its medial-orbital surface, the frontal pole, a strip of the lateral surface of the cerebral hemisphere along its superior border, and the anterior four-fifths of the corpus callosum. Deep branches, arising near the circle of Willis (proximal and distal to the anterior communicating artery) supply the anterior limb of the internal capsule, the inferior part of the head of the caudate nucleus, and the anterior part of the globus pallidus (see Figs. 34-5, 34-6, and 34-7). The largest of these deep branches is the artery of Heubner (“recurrent artery of Heubner”). This artery, which may, in fact, be up to four small vessels, shares its territory of supply with anteriorly placed lenticulostriate arteries that emanate from the middle cerebral artery. Strokes in this territory cause infarction of the head of the caudate. Most strokes are of the embolic variety, far less often atherosclerotic, and occasionally due to other processes such as vasospasm or vasculitis.
Figure 34-6.
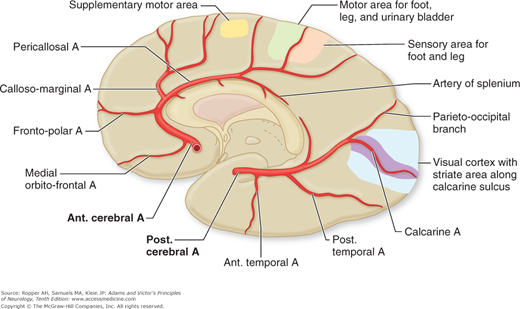
A. Diagram of the right cerebral hemisphere, medial aspect, showing the branches and distribution of the anterior cerebral artery and the principal regions of cerebral localization. Below is a list of the clinical manifestations of infarction in the territory of this artery and the corresponding regions of cerebral damage. Also shown is the course of the main branch of the posterior cerebral artery on the medial side of the hemisphere. Note: Hemianopia does not occur; transcortical aphasia occurs rarely (isolation of the language areas) (see Chap. 23). B. Axial diffusion-weighted MRI showing an acute ischemic infarction in the anterior cerebral artery territory.
Figure 34-7.
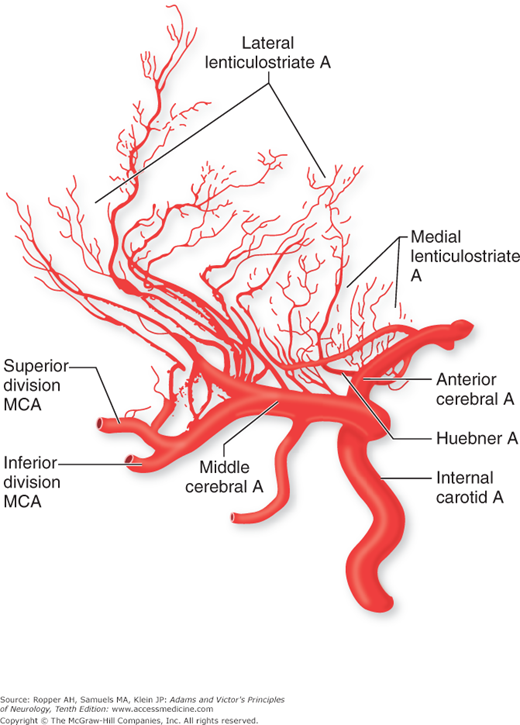
Corrosion preparations with plastics demonstrating penetrating branches of the anterior and middle cerebral arteries. The medial and lateral lenticulostriate arterioles are labeled, along with the recurrent artery of Heubner. (Reproduced by permission from Krayenbühl and Yasargil.)
The clinical picture of anterior cerebral artery stroke will depend on the location and size of the infarct, which, in turn, relates to the site of the occlusion (proximal or distal to the anterior communicating artery), the pattern of the circle of Willis, and the other ischemia-modifying factors mentioned earlier. Well-studied cases of infarction in the territory of the anterior cerebral artery are not numerous; hence the syndromes have not been completely elucidated (see Brust for a review of the literature and a description of developmental abnormalities of the artery).
Occlusion of the stem of the anterior cerebral artery, proximal to its connection with the anterior communicating artery (the A1 segment in neuroradiologic parlance) is usually well tolerated, because adequate collateral flow is provided by the anterior or cerebral artery of the opposite side. Maximal disturbance occurs when both arteries arise from one anterior cerebral stem, in which case there is infarction of the anterior and medial parts of both cerebral hemispheres resulting in paraplegia, incontinence, abulia and nonfluent aphasic symptoms, and frontal lobe personality changes (see Chap. 22). Occlusion of the anterior cerebral arteries is usually embolic, but atherothrombotic lesions are known and instances because of surgical occlusion by an aneurysm clip are well described.
Complete infarction as a result of occlusion of one anterior cerebral artery distal to the anterior communicating artery (A2 segment) results in a sensorimotor deficit of the opposite foot and leg and, to a lesser degree, of the shoulder and arm, with sparing of the hand and face (the distribution of which is shown in the MRI of Fig. 34-6). This is the complement of the superior division middle cerebral artery pattern. The motor disorder is more pronounced in the foot and leg than in the hip and thigh. Sensory loss, when it occurs, is mainly of the discriminative modalities but it may be mild or absent. The head and eyes may deviate to the side of the lesion. Urinary incontinence, a contralateral grasp reflex, and paratonic rigidity (gegenhalten) of the opposite limbs may be evident. With a left-sided occlusion, there may be a “sympathetic apraxia” of the left arm and leg or involuntary misdirected movements of the left arm (alien arm or hand), as described in Chaps. 3 and 22.
Language disturbances, particularly transcortical motor aphasia (see Chap. 23), may occur with anterior cerebral artery territory stroke. Alexander and Schmitt cited cases in which occlusions of the proximal artery (ACA), which included the Heubner artery, resulted in right hemiplegia (predominant in the leg) with grasping and groping responses of the right hand and buccofacial apraxia that were accompanied by a diminution or absence of spontaneous speech, agraphia, and a limited ability to name objects and compose word lists but with a striking preservation of the ability to repeat spoken and written sentences (i.e., transcortical motor aphasia). Disorders of behavior that may be overlooked in cases of anterior cerebral artery occlusion; they are abulia, or a slowness and lack of spontaneity in all reactions, muteness or a tendency to speak in whispers, and distractibility. Branch occlusions of the anterior cerebral artery produce only fragments of the total syndrome, usually a spastic weakness or associative sensory loss in the opposite foot and leg.
With occlusion of penetrating branches of the ACA, the anterior limb of the internal capsule and caudate is usually involved. In a series of 18 cases of unilateral caudate region infarcts collected by Caplan and associates, a transient hemiparesis was present in 13. Dysarthria and either abulia or agitation and hyperactivity were also common. Stuttering and language difficulty occurred with two of the left-sided lesions and visuospatial neglect with three of the right-sided ones. To what extent these symptoms were the result of a disturbance of neighboring structures is difficult to determine. With bilateral caudate infarctions, a syndrome of inattentiveness, abulia, forgetfulness, and sometimes agitation and psychosis was observed. Transitory choreoathetosis and other dyskinesias (we have seen two cases of ballismus) have also been attributed to ischemia of the caudate and anterior basal ganglia, occurring sometimes under conditions of prolonged standing and exercise (Caplan and Sergay; Margolin and Marsden).
This is a long, narrow artery that springs from the internal carotid, just above the origin of the posterior communicating artery. It supplies the internal segment of the globus pallidus and posterior limb of the internal capsule and several contiguous structures including (in most patients) the optic tract (see Fig. 34-5). It then penetrates the temporal horn of the lateral ventricle, where it supplies the choroid plexus and anastomoses with the posterior choroidal artery. This being a small caliber branch, most strokes in this territory are due to in situ atherosclerosis of the type that occurs in diabetics but occlusion of the orifice of the vessel by an embolus is possible and it is a known complication of clipping of an aneurysm at the upper reaches of the carotid artery.
Only a few complete clinicopathologic studies have been made of the distinctive syndrome caused by occlusion of this artery. It was found by Foix and colleagues to consist of contralateral hemiplegia, hemihypesthesia, and homonymous sectorial hemianopia (not reaching the vertical meridian of the visual fields) as a result of involvement of the posterior limb of the internal capsule and white matter posterolateral to it, through which the geniculocalcarine tract passes, and the lateral geniculate nucleus. This combination of extensive unilateral motor, sensory, and visual impairment in an individual with well-preserved language and cognition distinguishes this stroke syndrome from the more common ones involving the major cerebral arteries. Decroix and colleagues reported 16 cases (identified by CT) in which the lesion appeared to lie in the vascular territory of this artery. In most of their cases, the clinical syndrome fell short of what was expected on anatomic grounds or had additional features.
With right-sided lesions, there may be a left spatial neglect and constructional apraxia; slight disorders of speech and language may accompany left-sided lesions. Hupperts and colleagues have discussed the controversy regarding the effects of occlusion of the artery and in particular the variability of its supply to the posterior paraventricular area of the corona radiata and adjacent regions. They concluded, also from a survey of CT images, that there was no uniform syndrome attributable to occlusion of the vessel and that in most cases its territory of supply was overlapped by small surrounding vessels. It may be remembered that for a time, in order to abolish the tremor and rigidity of unilateral Parkinson disease, the anterior choroidal artery was being surgically ligated without these other effects having been produced.
In approximately 70 percent of individuals, both posterior cerebral arteries are formed by the bifurcation of the basilar artery and thin posterior communicating arteries join this system to the internal carotid arteries. In 20 to 25 percent, one posterior cerebral artery arises from the basilar in the usual way, but the other arises from the internal carotid, a persistent fetal pattern of circulation; fewer than 5 percent have the unusual configuration in which both arise from the corresponding carotid arteries. Most strokes in this territory are embolic in origin but some individuals are predisposed to atherosclerosis in the proximal posterior cerebral artery. There is also the possibility of ischemia in this territory from occlusion of more proximal vessels, particularly the basilar artery, or infraction in the distal territory of the vessel as a result of global failure of cerebral perfusion, as in severe hypotension.
The configuration and branches of the proximal segment of the posterior cerebral artery (P1 segment) are illustrated in Figs. 34-6, 34-8, and 34-9. The interpeduncular branches, which arise just above the basilar bifurcation, supply the red nuclei, the substantia nigra bilaterally, medial parts of the cerebral peduncles, oculomotor and trochlear nuclei and nerves, reticular substance of the upper brainstem, decussation of the superior cerebellar peduncles, medial longitudinal fasciculi, and medial lemnisci. The P1 portion of the posterior cerebral artery, giving rise to the interpeduncular branches (that portion between the bifurcation of the basilar artery and the ostium of the posterior communicating artery), is also referred to as the mesencephalic artery or the basilar communicating artery. As pointed out by Percheron (whose name is often applied to the largest of these vessels), the arterial configuration of the paramedian mesencephalic arteries varies considerably: in some cases, two small vessels arise symmetrically, one from each side; in others, a single artery arises from one posterior cerebral stem (proximal P1), which then bifurcates. In the latter case, one posterior cerebral stem supplies the medial thalamic territories on both sides, and an occlusion of this artery or one common paramedian trunk produces a bilateral butterfly-shaped lesion in the medial parts of the diencephalon. These are elaborated and illustrated by Castaigne et al.
Figure 34-8.
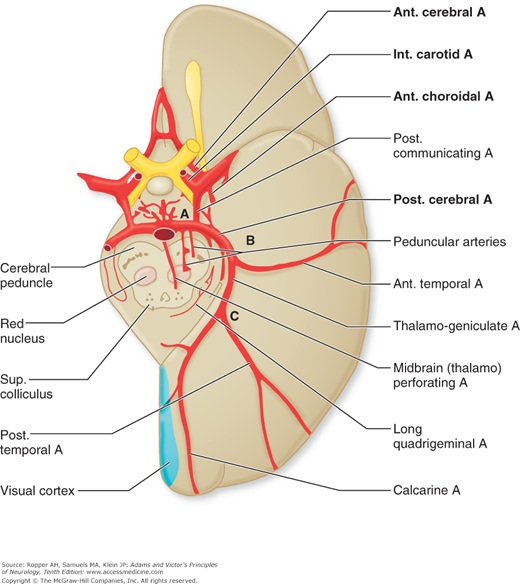
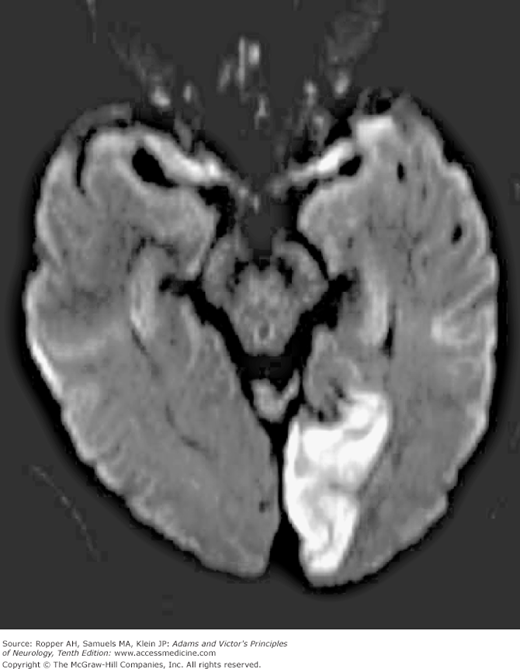
A. Inferior aspect of the left hemisphere showing the branches and distribution of the posterior cerebral artery and the principal anatomic structures supplied. The vessel is considered from the perspective of its proximal and distal territories. Listed below are the clinical manifestations produced by infarction in these territories and the corresponding regions of damage. Tremor in repose has been omitted because of the uncertainty of its occurrence in the posterior cerebral artery syndrome. Peduncular hallucinosis may occur in thalamic-subthalamic ischemic lesions, but the exact location of the lesion is unknown. B. Axial diffusion-weighted MRI showing an acute ischemic infarction in the posterior cerebral artery territory.
Figure 34-9.

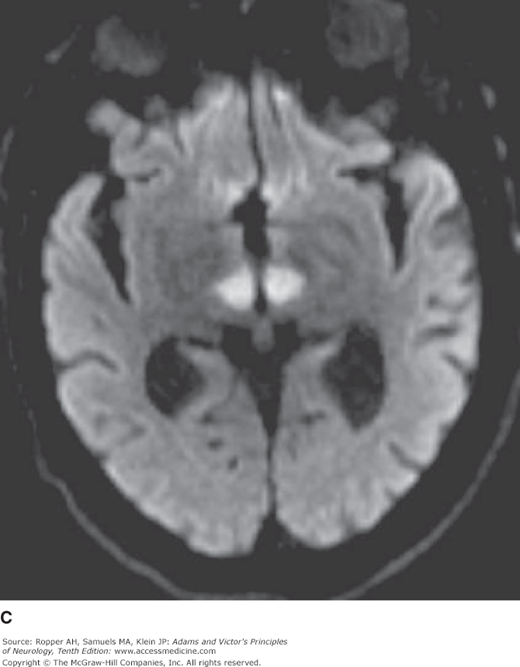
The posterior cerebral and basilar arteries. A. The terminus of the basilar artery and branches originating from the P1 through P3 segments. (Reproduced by permission from Stroke 34:2264, 2003.) B. Lateral view of the brain showing the branches of the posterior cerebral artery. (Reproduced by permission from Krayenbühl and Yasargil.) C. Axial diffusion-weighted MRI showing an acute ischemic infarction due to occlusion of an artery of Percheron, an anatomic variant, in which an azygos paramedian artery supplies both sides of the posterior-medial thalamus.
The thalamoperforate branches (also called paramedian thalamic arteries) arise slightly more distally from the stem, nearer the junction of the posterior cerebral and posterior communicating arteries (P2 segment of the artery) and supply the inferior, medial, and anterior parts of the thalamus. The thalamogeniculate branches arise still more distally, opposite the lateral geniculate body, and supply the geniculate body and the central and posterior parts of the thalamus. Medial branches emerging from the posterior cerebral artery as it encircles the midbrain, supply the lateral part of the cerebral peduncle, lateral tegmentum and corpora quadrigemina, and pineal gland. Posterior choroidal branches run to the posterosuperior thalamus, choroid plexus, posterior parts of the hippocampus, and psalterium (decussation of deep white matter fornices).
Most importantly, the terminal or cortical branches of the posterior cerebral artery supply the inferomedial part of the temporal lobe and the medial occipital lobe, including the lingula, cuneus, precuneus, and visual Brodmann areas 17, 18, and 19 (see Figs. 34-6, 34-8, and 34-9).
Occlusion of the posterior cerebral artery produces a greater variety of clinical effects than occlusion of any other artery because both the upper brainstem, which is replete with important structures, and the inferomedial parts of the temporal and occipital lobes lie within its supply. The site of the occlusion and the arrangement of the circle of Willis will, in large measure, determine the location and extent of the resulting infarct. For example, occlusion proximal to the posterior communicating artery may be asymptomatic or have only transitory effects if the collateral flow is adequate from the opposite posterior cerebral vessel (see Fig. 34-8; see also Fig. 34-9). Even distal to the posterior communicating artery, an occlusion may cause relatively little damage if the collateral flow through border-zone collaterals from anterior and middle cerebral arteries is sufficient.
In the series of posterior cerebral artery strokes studied by Milandre and coworkers, the causes were, in general, similar to those of strokes in other vascular territories except that there was a higher incidence of atherosclerotic occlusion (35 patients) in contrast to cardioembolic types (15 patients). Two of their stroke cases were attributed to migraine. Our experience has differed in that the proportion of presumed embolic occlusions has been far greater than that of other causes.
For convenience of exposition, it is helpful to divide the posterior cerebral artery syndromes into three groups: (1) proximal (involving interpeduncular, thalamic perforant, and thalamogeniculate branches), (2) cortical (inferior temporal and medial occipital), and (3) bilateral.
Figure 34-10.
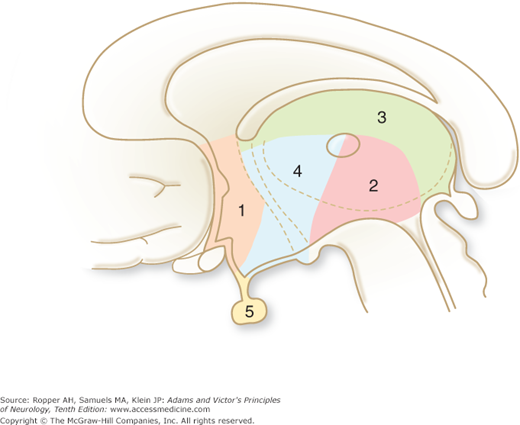
Diagram of the regions of blood supply of the diencephalon. Distribution of the (1) anterior cerebral artery, (2) posterior cerebral artery, (3) anterior and posterior choroidal arteries, (4) posterior communicating artery, and (5) internal carotid artery. (Reproduced by permission from Krayenbühl and Yasargil.)
The thalamic syndrome of Dejerine and Roussy (see also Chap. 8) follows infarction of the sensory relay nuclei in the thalamus, the result of occlusion of thalamogeniculate branches. Occlusion of the small vessels supplying these territories from in situ atherothrombosis or embolic occlusion of the posterior cerebral artery is the most common cause. There is both a deep and cutaneous sensory loss, usually severe in degree, of the opposite side of the body, including the trunk and face, sometimes accompanied by a transitory hemiparesis. A homonymous hemianopia may be conjoined. In some instances there is a dissociated sensory loss—pain and thermal sensation being more affected than touch, vibration, and position—or only one part of the body may be anesthetic. The characteristic feature is always sensory loss that includes the entire hemibody up to the midline. After an interval, sensation begins to return, and the patient may develop pain, paresthesia, and hyperpathia in the affected parts. The painful paresthetic syndrome may persist for years. There may also be distortion of taste, athetotic posturing of the hand, and alteration of mood. Mania and depression have occasionally been observed with infarction of the diencephalon and adjacent structures, but the data are usually incomplete.
Central midbrain and subthalamic syndromes are a result of occlusion of the interpeduncular branches of the posterior cerebral artery. The clinical syndromes include palsies of vertical gaze, stupor, or coma. Syndromes of the paramedian arteries, including the proximal posterior cerebral artery, have as their main feature a third-nerve palsy combined with contralateral hemiplegia (Weber syndrome), contralateral ataxic tremor (Claude syndrome), or homolateral ataxia, hemiplegia with contralateral third-nerve palsy (Benedikt syndrome), as summarized in Table 34-5.
EPONYMb | SITE | CRANIAL NERVES INVOLVED | TRACTS INVOLVED | SIGNS | USUAL CAUSE |
|---|---|---|---|---|---|
Weber syndrome | Base of midbrain | III | Corticospinal tract | Oculomotor palsy with crossed hemiplegia | Vascular occlusion, tumor, aneurysm |
Claude syndrome | Tegmentum of midbrain | III | Red nucleus, superior cerebellar peduncles after decussation | Oculomotor palsy with contralateral cerebellar ataxia and tremor | Vascular occlusion, tumor, aneurysm |
Benedikt syndrome | Tegmentum of midbrain | III | Red nucleus, corticospinal tract, and superior cerebellar peduncles after decussation | Oculomotor palsy with contralateral cerebellar ataxia, tremor, and corticospinal signs, may have choreoathetosis | Infarct, hemorrhage, tuberculoma, tumor |
Nothnagel syndrome | Tectum of midbrain | Unilateral or bilateral III | Superior cerebellar peduncles | Ocular palsies (IV), paralysis of gaze, nystagmus, and ataxia | Tumor |
Parinaud syndrome | Dorsal midbrain | Supranuclear mechanism for upward gaze and other structures in periaqueductal gray matter | Paralysis of upward gaze and accommodation; fixed pupils | Pinealoma and other lesions of dorsal midbrain, hydrocephalus | |
Millard-Gubler syndrome and Raymond-Foville syndrome | Base of pons | VII and often VI | Corticospinal tract | Facial and abducens palsy and contralateral hemiplegia; sometimes gaze palsy to side of lesion | Infarct or tumor |
Avellis syndrome | Tegmentum of medulla | X | Spinothalamic tract; sometimes descending pupillary fibers, with Bernard-Horner syndrome | Paralysis of soft palate and vocal cord and contralateral hemianesthesia | Infarct or tumor |
Jackson syndrome | Tegmentum of medulla | X, XII | Corticospinal tract | Avellis syndrome plus ipsilateral tongue paralysis | Infarct or tumor |
Wallenberg syndrome | Lateral tegmentum of medulla | Spinal V, IX, X, XI | Lateral spinothalamic tract | Ipsilateral V, IX, X, XI palsy, Horner syndrome, and cerebellar ataxia; contralateral loss of pain and temperature sense | Occlusion of vertebral or posterior–inferior cerebellar artery |
Descending pupillodilator fibers | |||||
Spinocerebellar and olivocerebellar tracts |
Anteromedial-inferior thalamic syndromes follow occlusion of the thalamoperforant branches. Here the most common effect is an extrapyramidal movement disorder (hemiballismus or hemichoreoathetosis or less often, asterixis). Deep sensory loss, hemiataxia, or tremor may be added in various combinations. Hemiballismus is usually a result of occlusion of a small branch to the subthalamic nucleus (of Luys) or its connections with the pallidum. Occlusion of the paramedian thalamic branches to the mediodorsal nucleus is a recognized cause of an amnesic (Korsakoff) syndrome; this simulates but is less common than infarction of the hippocampi from occlusion of the medial temporal branch of the posterior cerebral artery as noted below.
Occlusion of branches to the temporal and occipital lobes gives rise to a homonymous hemianopia as a result of involvement of the primary visual receptive areas (calcarine or striate cortex) or of the converging geniculocalcarine fibers. The hemianopia may be incomplete and involve the upper quadrants of the visual fields more than the lower ones (see Chap. 13). Macular, or central, vision is often spared because of collateral blood supply of the occipital pole from distal branches of the middle (or anterior) cerebral arteries. Other features seen in a few instances are visual hallucinations in the blind parts of the visual fields (Cogan) or metamorphopsia and palinopsia (Brust and Behrens). Occipital infarcts of the dominant hemisphere may cause alexia without agraphia, anomia (amnesic aphasia), a variety of visual agnosias, and rarely some degree of impaired memory. The anomias, when they occur, are most severe for colors, but the naming of other visually presented material such as pictures, mathematical symbols, and manipulable objects may also be impaired. The patient may treat objects as familiar—that is, describe their functions and use them correctly—but be unable to name them. Color anomia (a form of “central achromatopsia”) and amnesic aphasia are more often present in this syndrome than is alexia. The defect in retentive memory is of varying severity and may or may not improve with the passage of time. These syndromes are described in Chaps. 22 and 23.
A complete proximal arterial occlusion leads to a syndrome that combines cortical and anterior-proximal syndromes in part or totally. As mentioned, the vascular lesion may be either an embolus or an atherosclerotic thrombus.
These occur as a result of successive infarctions or from a single embolic or thrombotic occlusion of the upper basilar artery, especially if the posterior communicating arteries are unusually small or absent, or from global failure of circulation.
Bilateral lesions of the occipital lobes, if extensive, cause “cortical blindness” that is essentially bilateral homonymous hemianopia, sometimes accompanied by unformed visual hallucinations. The pupillary reflexes are preserved and the optic discs appear normal. Sometimes the patient is unaware of being blind and denies the problem even when it is pointed out to him (Anton syndrome). More frequently, the lesions are incomplete, and a sector of the vision, usually on one side, remains intact. When the visual remnant is small, vision may seemingly fluctuate from moment to moment as the patient attempts to capture the image in the island of intact vision, in which case hysteria may be incorrectly inferred. In bilateral lesions confined to the occipital poles, there may be a loss of central vision only (homonymous central scotomas). With more anteriorly placed lesions of the occipital pole, there may be homonymous paracentral scotomas, or the occipital poles may be spared, leaving the patient with only central vision. Horizontal or altitudinal field defects are usually a result of similar restricted lesions affecting the upper or lower banks of the calcarine sulci. The Balint syndrome (see Chap. 22) is an effect of bilateral occipitoparietal border-zone lesions.
With bilateral lesions that involve the inferomedial portions of the temporal lobes, including the hippocampi and their associated structures, the impairment of memory may be severe, causing the Korsakoff amnesic state. In several of our patients, a solely left-sided infarction of the inferomedial temporal lobe impaired retentive memory. Bilateral mesiotemporal-occipital lesions also cause a lack of recognition of faces (prosopagnosia). These and other effects of temporal and occipital lesions are discussed in Chaps. 13 and 22.
The vertebral arteries are the chief arteries of the medulla; each supplies the lower three-fourths of the pyramid, the medial lemniscus, all or nearly all of the retroolivary (lateral medullary) region, the restiform body, and the posteroinferior part of the cerebellar hemisphere through the posterior inferior cerebellar arteries (Fig. 34-11). The relative sizes of the vertebral arteries vary considerably, and in approximately 10 percent of cases, one vessel is so small that the other is essentially the only artery of supply to the brainstem. In the latter cases, if there is no collateral flow from the carotid system via the circle of Willis, occlusion of the one functional vertebral artery is equivalent to occlusion of the basilar artery (see below). The posteroinferior cerebellar artery (PICA) is usually a branch of the vertebral artery but can have a common origin and form a loop with the anteroinferior cerebellar artery (AICA) from the basilar artery. It is necessary to keep these anatomic variations in mind in considering the effects of vertebral artery occlusion.
Figure 34-11.
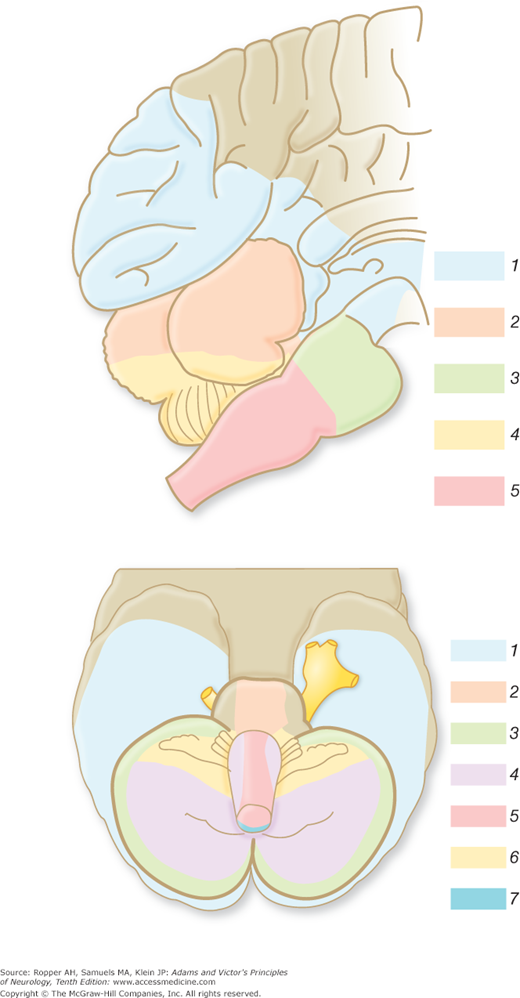
Regions supplied by the posterior segment of the circle of Willis, lateral view (A) and basal view (B). A. (1) Posterior cerebral artery; (2) superior cerebellar artery; (3) basilar artery and superior cerebellar artery; (4) posterior inferior cerebellar artery; (5) vertebral artery (posterior inferior cerebellar artery, anterior spinal artery, posterior spinal artery). B. (1) Posterior cerebral artery; (2) superior cerebellar artery; (3) paramedian branches of the basilar artery and spinal artery; (4) posterior inferior cerebellar artery; (5) vertebral artery; (6) anterior inferior cerebellar artery; (7) dorsal spinal artery. (Reproduced by permission from Krayenbühl and Yasargil.)
The vertebral arteries are most often occluded by atherothrombosis in their intracranial portion. Because the vertebral arteries have a long extracranial course and pass through the transverse processes of C6 to C1 vertebrae before entering the cranial cavity, one might expect them to be subject to trauma, spondylotic compression, and a variety of other vertebral diseases. With the exception of arterial dissection, in our experience the other causes of vascular occlusion happen only infrequently. We rarely see convincing examples of spondylotic occlusion but several such cases have been reported. Extreme extension of the neck, as experienced by women who are having their hair washed in beauty salons, or during yoga positions, may give rise to transient symptoms in the territory of the vertebral artery.
Dissection of the vertebral artery by contrast is well described; it declares itself by cervicooccipital pain and deficits of brainstem function. One’s attention is drawn to the diagnosis of vertebral dissection where there have been vigorous and protracted bouts of coughing or trauma to the neck or head. Dissection of the extracranial vessels is discussed in more detail further on. Examples of posterior circulation stroke in children have been reported in association with odontoid hypoplasia and other atlantoaxial dislocations, causing the vertebral arteries to be stretched or kinked in their course through the transverse processes of C1-C2 (Phillips et al).
The results of vertebral artery occlusion are quite variable. If the occlusion of the vertebral artery is so situated as to block the posterior inferior cerebellar artery supplying the lateral medulla and inferior cerebellum (PICA), a characteristic syndrome results with vertigo being a prominent symptom (see “Lateral Medullary Syndrome” described below). If the subclavian artery is blocked proximal to the origin of the left vertebral artery, exercise of the arm on that side may draw blood from the right vertebral and basilar arteries, retrograde down the left vertebral and into the distal left subclavian artery—sometimes resulting in the symptoms of basilar insufficiency. This phenomenon, described in 1961 by Reivich and colleagues, was referred to by Fisher (1961) as the subclavian steal. Its most notable features are vertigo and other brainstem signs coupled with transient weakness on exercise of the left arm. There may also be headache and claudication or pain of the arm. In an unusual configuration in which one vertebral artery is occluded just proximal to the origin of its PICA branch, and the opposite vertebral artery is open and sufficient in size, there may be no symptoms because the PICA is still filled by retrograde flow through its vertebral artery.
Less often, occlusion of the vertebral artery or one of its medial branches produces an infarct that involves the medullary pyramid, the medial lemniscus, and the emergent hypoglossal fibers; the resultant syndrome consists of a contralateral paralysis of arm and leg (with sparing of the face), contralateral loss of position and vibration sense, and ipsilateral paralysis and later atrophy of the tongue. This is the medial medullary syndrome (Fig. 34-12). A more limited lesion, from occlusion of one spinal artery arising from the vertebral artery, gives rise to a contralateral hemiplegia (rarely a quadriplegia) that spares the face. When the vertebral branch to the anterior spinal artery is blocked, flow from the other (corresponding) branch is usually sufficient to prevent infarction of the cervical cord, but we and others have described solely pyramidal infarction with hemiplegia or quadriplegia that spares the face (Ropper et al). Occlusion of a vertebral artery low in the neck is usually compensated by anastomotic flow to the upper part of the artery via the thyrocervical, deep cervical, and occipital arteries or by reflux from the circle of Willis.
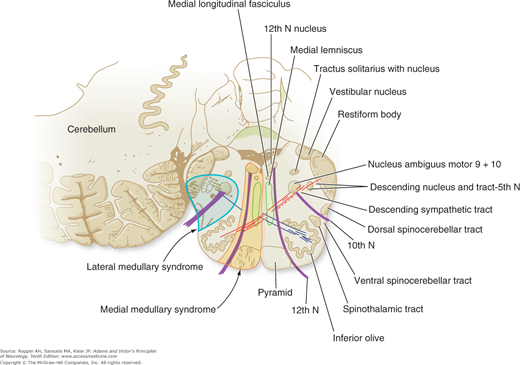
Known also as the Wallenberg syndrome (who described a case in 1895), this common stroke is produced by infarction of a wedge of lateral medulla lying posterior to the inferior olivary nucleus (see Fig. 34-12). The complete syndrome, as outlined by Fisher and colleagues (1961), comprises (a) symptoms derived from the vestibular nuclei (vertigo, nystagmus, oscillopsia, vomiting); (b) spinothalamic tract (contralateral or, less often, ipsilateral impairment of pain and thermal sense over half the body); (c) descending sympathetic tract (ipsilateral Horner syndrome—miosis, ptosis, decreased sweating); (d) issuing fibers of the ninth and tenth nerves (hoarseness, dysphagia, hiccough, ipsilateral paralysis of the palate and vocal cord, diminished gag reflex); (e) utricular nucleus (vertical diplopia and illusion of tilting of vision and rotation of the vertical meridian, rarely so severe as to produce upside down vision); (f) olivocerebellar, spinocerebellar fibers, restiform body and inferior cerebellum (ipsilateral ataxia of limbs, falling or toppling to the ipsilateral side, and the sensation of lateropulsion); (g) descending tract and nucleus of the fifth nerve (pain, burning, and impaired sensation over ipsilateral half of the face); (h) nucleus and tractus solitarius (loss of taste); and rarely, (i) cuneate and gracile nuclei (numbness of ipsilateral limbs). Fragmentary syndromes are more frequent, especially at the onset of the stroke. These subsyndromes may consist of vertigo and ptosis, toppling and vertical diplopia, hoarseness and disequilibrium, or other combinations short of the entire syndrome. While vertigo is the most frequent feature alone, it is not usually an indication of lateral medullary infarction. The smallest infarction we have studied gave rise only to symptoms of lateropulsion and mild ipsilateral limb ataxia.
The eye signs of lateral medullary infarction are also varied and quite interesting. There is often a fragment of an internuclear ophthalmoplegia or a skew deviation (the globe on the affected side usually being higher). Direction-changing nystagmus (with different head positions) is a useful feature that suggests labyrinthine disease from brainstem forms of nystagmus, but infarction of the vestibular nucleus as part of the lateral medullary syndrome may also produce this sign (see Chap. 15).
The entire lateral medullary syndrome, one of the most striking in neurology, is almost always caused by infarction, with only a small number of cases being the result of hemorrhage, demyelination, or tumor. Although it has traditionally been attributed to occlusion in the course of the PICA, as mentioned earlier, careful studies have shown that in 8 of 10 cases it is the vertebral artery that is occluded by atherothrombosis; in the remainder, either the posterior inferior cerebellar artery or one of the lateral medullary arteries is occluded. Embolism to the PICA is a less-frequent cause. The inferior cerebellum is usually affected, sometimes in isolation if the embolus travels distal to the origin of PICA, causing a cerebellar infarct with attendant vomiting, vertigo, and ataxia often with occipitonuchal headache.
In recent years, we have had experience with patients who initially have considerable recovery in the first days and weeks, but experience sudden and unexpected death from respiratory or cardiac arrest, even in the absence of cerebellar swelling or basilar artery thrombosis. Cases of this nature have been reviewed by Norrving and Cronqvist. The related and important issue of cerebellar swelling after vertebral artery or PICA occlusion and the need for surgical decompression is discussed later in the chapter.
The branches of the basilar artery may be instructively grouped as follows: (1) paramedian, 7 to 10 pairs, supplying a wedge of pons on either side of the midline; (2) short circumferential, 5 to 7 pairs in number, supplying the lateral two-thirds of the pons and the middle and superior cerebellar peduncles; (3) long circumferential, 2 on each side (the superior and anterior inferior cerebellar arteries), which run laterally around the pons to reach the cerebellar hemispheres (see Figs. 34-11 and 34-13 through 15); and (4) several paramedian (interpeduncular) branches at the bifurcation of the basilar artery and origins of the posterior cerebral arteries supplying the high midbrain and medial subthalamic regions. These interpeduncular and other short proximal branches of the posterior cerebral artery were described earlier in the chapter.
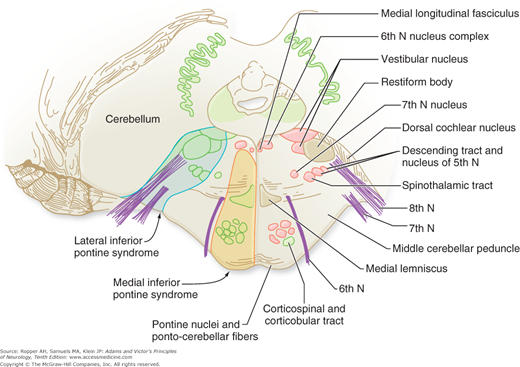
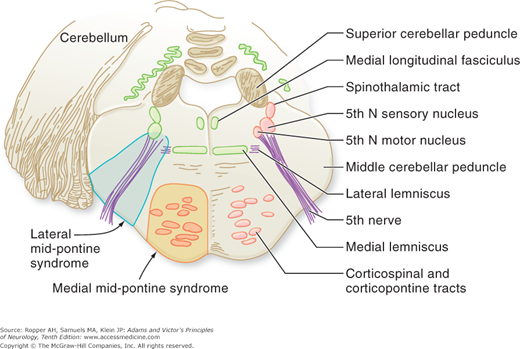
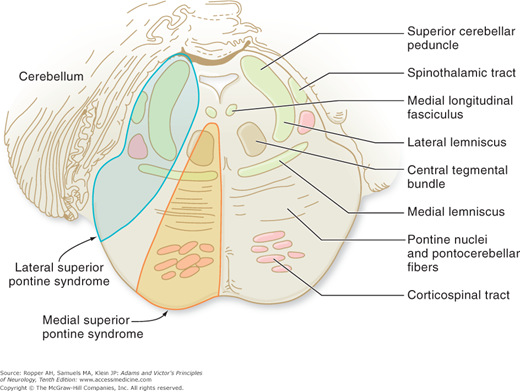
Basilar artery occlusion, typically because of local thrombosis that is superimposed on a preexisting atherosclerotic plaque, can arise in several ways: (1) occlusion of the basilar artery itself, usually in the lower or middle third at the site of an atherosclerotic plaque; (2) occlusion of both vertebral arteries, which produces the equivalent of basilar artery occlusion if the circle of Willis is inadequate; and (3) occlusion of a single vertebral artery when it is the only one of adequate size. When there is embolism, the clot usually lodges at the terminal bifurcation of the basilar (“top-of-the-basilar syndrome”) or in one of the posterior cerebral arteries, as the clot, if small enough to pass through the vertebral artery, easily traverses the length of the basilar artery, which is of greater diameter than either vertebral artery. Also, thrombosis may involve a branch of the basilar artery rather than the trunk (basilar branch occlusion).
The syndrome of basilar artery occlusion, as delineated by Kubik and Adams, reflects the involvement of a large number of bilateral structures: corticospinal and corticobulbar tracts; cerebellum, middle and superior cerebellar peduncles; medial and lateral lemnisci; spinothalamic tracts; medial longitudinal fasciculi; pontine nuclei; vestibular and cochlear nuclei; descending hypothalamospinal sympathetic fibers; and the third through eighth cranial nerves (the nuclei and their segments within the brainstem). Thus the complete syndrome comprises bilateral long tract signs (sensory and motor) with variable cerebellar, cranial nerve, and other segmental abnormalities of the brainstem.
Another important syndrome, the result of occlusion of the distal end of the basilar artery, consists of coma from infarction of the high midbrain reticular activating system. This “top of the basilar” artery occlusion is most often embolic and characterized by transient loss of consciousness, oculomotor disturbances (roving eye movements or eyes looking downward and inward with inability to reflexly elicit upward movements), hemianopia, bilateral ptosis, and pupillary enlargement with preserved reaction to light. Spontaneous recanalization of the vessel is frequent, but occurs in a delayed fashion, after the infarct has been established. MRI demonstrates a characteristic pattern of central midbrain, bilateral posterior thalamic, and, more variably, uni- or bilateral posterior territory infarction as discussed below.
Yet another configuration, the result of occlusion of the midbasilar artery, gives rise to the locked-in syndrome, in which the patient is mute and quadriplegic but conscious, reflecting interruption of descending motor pathways in the base of the pons but sparing of the reticular activating system (“locked-in” syndrome; see Chap. 17). Horizontal eye movements are obliterated but vertical ones and some ability to elevate the eyelids are spared. The pupils become extremely small but retain some reaction to light. Midbasilar disease may also cause coma if the posterior communicating arteries are inadequate to perfuse the distal basilar artery territory.
In the presence of the full syndrome, it is usually not difficult to make the correct diagnosis. The aim should be, however, to recognize basilar insufficiency long before the stage of total deficit has been reached. The early manifestations (in the form of TIAs) occur in many combinations, described in detail further on.
Occlusion of branches at the bifurcation (top) of the basilar artery results in a remarkable number of complex syndromes that include, in various combinations, somnolence or coma, memory defects, akinetic mutism, visual hallucinations, ptosis, disorders of ocular movement (convergence spasm, paralysis of vertical gaze, retraction nystagmus, pseudoabducens palsy, retraction of upper eyelids, skew deviation of the eyes), an agitated confusional state, and visual field defects (see Fig. 34-9). These have been summarized by Petit and coworkers and Castaigne and associates and categorized as paramedian thalamic, subthalamic, and midbrain syndromes, and by Caplan as parts of the “top of the basilar” syndrome. Limited, small infarctions on one side of the brainstem are usually due to occlusion of small penetrating vessels that originate in the basilar artery. Emboli coursing through the basilar artery can also occlude the mouths of several small penetrating vessels and cause larger infractions. A larger infarction in the territory of one circumferential vessel is usually due to an embolus but may also result from an atherosclerotic plaque in the parent basilar artery. The distinction is often made by the rapidity of onset and the presence of risk factors such as atrial fibrillation for embolus or diabetes and hypertension for small vessel occlusion.
The main signs of occlusion of the superior cerebellar artery, the most rostral circumferential branch of the basilar, are ipsilateral cerebellar ataxia of the limbs (referable to middle and superior cerebellar peduncles); nausea and vomiting; slurred speech; and loss of pain and thermal sensation over the opposite side of the body (spinothalamic tract). Partial deafness, static tremor of the ipsilateral upper extremity, an ipsilateral Horner syndrome, and palatal myoclonus have also been reported.
With occlusion of the anterior inferior cerebellar artery (AICA), the extent of the infarct is extremely variable, as the size of this artery and the territory it supplies vary inversely with the size and territory of supply of the PICA. The principal findings are vertigo, vomiting, nystagmus, tinnitus, and sometimes unilateral deafness; facial weakness; ipsilateral cerebellar ataxia (inferior or middle cerebellar peduncle); an ipsilateral Horner syndrome and paresis of conjugate lateral gaze; and contralateral loss of pain and temperature sense of the arm, trunk, and leg (lateral spinothalamic tract) as shown in Fig. 34-14. The tinnitus, if present at all, may be overwhelming, called “screaming” by some of our patients. If the occlusion is close to the origin of the artery, the corticospinal fibers may also be involved, producing a hemiplegia; if distal, there may be cochlear and labyrinthine infarction. Cerebellar swelling did not occur with AICA territory infarction in the 20 collected by Amarenco and Hauw but it has been a more common occurrence in our material.
The most characteristic manifestation of all these basilar branch strokes is the “crossed” cranial nerve and long tract sensory or motor deficit reflecting a unilateral segmented infarction of the brainstem. These syndromes, which may involve any of cranial nerves III through XII, are listed in Table 34-5. Although the finding of bilateral neurologic signs strongly suggests brainstem involvement, these syndromes make it apparent that in many instances of infarction within the basilar territory, the signs are limited to one side of the body.
In the absence of one of the localizing crossed cranial nerve signs it is often not possible to distinguish a hemiplegia of pontine origin from one of deep cerebral origin on the basis of motor signs alone. In both, the face, arm, hand, leg, and foot are affected because of the compression of the descending motor fibers into a small segmental region. With brainstem lesions as with cerebral ones, a flaccid paralysis gives way to spasticity after a few days or weeks, and there is no satisfactory explanation for the occurrence in some cases of spasticity from the onset of the stroke. There is also often a combined hemiparesis and ataxia of the limbs on the same side. With a hemiplegia of pontine origin, however, the eyes may deviate to the side of the paralysis, i.e., the opposite of what occurs with supratentorial lesions. The pattern of sensory disturbance may also be helpful. A dissociated sensory deficit over the ipsilateral face and contralateral half of the body usually indicates a lesion in the lower brainstem, while a hemisensory loss including the face and involving all modalities indicates a lesion in the upper brainstem, in the thalamus, or deep in the white matter of the parietal lobe. When position sense, two-point discrimination, and tactile localization are affected relatively more than pain or thermal and tactile sense, a cerebral lesion is suggested; the converse indicates a brainstem localization. Bilateral motor and sensory signs are almost certain evidence that the lesion lies in the brainstem. When hemiplegia or hemiparesis and sensory loss are coextensive, the lesion usually lays supratentorially. Additional manifestations that strongly favor a brainstem site are rotational dizziness, diplopia, cerebellar ataxia, a Horner syndrome, and deafness. The numerous brainstem syndromes illustrate the important point that the cerebellar pathways, spinothalamic tract, trigeminal nucleus, and sympathetic fibers can be involved at different rostral-caudal levels so that “neighboring” phenomena are required to identify the exact site of the infarction.
A myriad of proper names have been applied to the brainstem syndromes, as noted in Tables 34-5 and 47-1. Many of them were originally described in relation to tumors, trauma, and other nonvascular diseases. The diagnosis of vascular disorders in this region of the brain is not greatly facilitated by knowledge of these eponymic syndromes; it is much more profitable to be closely familiar with the anatomy of the brainstem. To recapitulate, the principal syndromes to be recognized are the full basilar, vertebral–PICA, posteroinferior cerebellar, anteroinferior cerebellar, superior cerebellar, pontomedullary, and medial medullary. Figures 34-12, 34-13, 34-14, and 34-15, supplied by C.M. Fisher and used in all previous editions of this book, present both medial and lateral syndromes at several levels of the medulla and pons. Other syndromes can usually be identified as fragments or combinations of the major ones.
As one might surmise, small penetrating branches of the cerebral arteries may become occluded, and the resulting infarcts may be so small or so situated as to cause no symptoms whatsoever. As the softened tissue is removed by macrophages, a small cavity, or lacune, remains. Early in the twentieth century, Pierre Marie referred to the condition as état lacunaire (the lesions were first described by Durant-Fardel in 1843). He distinguished these lesions from a fine loosening of tissue around thickened small vessels that enter the anterior and posterior perforated spaces, a change to which he gave the name état criblé (cribriform change). Pathologists have not always agreed on these distinctions, but we have adhered to the view of Fisher and Adams that lacunes are usually caused by occlusion of small arteries, 50 to 200 microns in diameter, and the cribriform state, to mere thickening of vessels and fraying of the surrounding tissue—i.e., dilated perivascular spaces (Virchow-Robin spaces) that do not have a corresponding neurologic disease.
In almost all clinical and pathologic material, there has been a strong relationship between the lacunar state and chronic hypertension, but also diabetes and hyperlipidemia. Sacco and colleagues (1991), in a population-based study in Rochester, Minnesota, found that 81 percent of patients with lacunar infarctions were also hypertensive. There appear to be three mechanisms for lacunar infarction but variants of atherothrombosis are foremost. The first, and the one most characteristically tied to lacunes, is a local type of fibrohyalinoid arteriolar sclerosis that involves the orifice or proximal part of a small penetrating blood vessel (lipohyalinosis as described below). The second is atherosclerosis of a large trunk vessel that occludes the origin of these same small vessels. This is prone to involve several adjacent vessels and cause, at times, larger lacunes or the atherosclerosis extends from a trunk vessel into a smaller one. Third is the entry of small embolic material into one of the vessels. The relative frequency of these three pathologies is not known, but the first seems to be most common and occurs without pathologic change in the trunk vessel of the circle of Willis whereas the embolic type is least frequent. When Fisher (1975) examined a series of such lesions in serial sections, from a basal parent artery up to and through the lacune, he was able to confirm a lipohyalin degeneration of the vessel wall and occlusion in the initial course of small vessels in most cases. In some, lipohyalinotic changes had resulted in false aneurysm formation, resembling the Charcot-Bouchard aneurysms, another hypertension-related change that underlies brain hemorrhage (see further on). In a series of 1,042 consecutive adults whose brains were examined postmortem, Fisher (1965b) observed one or more lacunes in 11 percent. He found 4 to 6 and sometimes up to 10 to 15 lacunes in any given brain specimen. In recent years, better treatment of hypertension has greatly reduced this number and the overall frequency of lacunar infarction, at least as judged by MRI.
Lacunes are situated, in descending order of frequency, in the putamen and caudate nuclei, thalamus, basis pontis, internal capsule, and deep in the central hemispheral white matter. The cavities range from 3 to 15 mm in diameter, and whether they cause symptoms depends entirely on their location.
Lacunar strokes tend to evolve quickly but not typically as suddenly as an embolus, for example. These clinical aspects are discussed extensively in the earlier section “Lacunar TIA.” In broad terms, the essential feature of these deep strokes is the uniform and striking absence of cortical deficits; that is, seizures, aphasia or amnesia (except in limited circumstances of small thalamic infarction), agnosia, apraxia, dysgraphia, alexia, and, a large number of similar cognitive changes that only begin to appear when there are multiple deep strokes and a dementia of the type discussed in Chap. 21. Furthermore, because of the small size of the strokes, certain clinical syndromes that might be expected to result from deep lesions, such as hemianopia, also do not occur. Fisher, in several papers, has delineated the most frequent symptomatic forms of lacunar stroke:
Pure motor hemiplegia
Pure sensory stroke
Clumsy hand–dysarthria
Ipsilateral hemiparesis–ataxia
A lacune in the territory of a lenticulostriate artery, i.e., in the internal capsule or adjacent corona radiata, usually causes a highly characteristic syndrome of pure motor hemiplegia involving the opposite face, arm, hand, leg, and foot in approximately equal measure. A lacune situated in the ventral pons causes an identical syndrome (Fig. 34-16). In both cases, the lacunar syndrome is identified as much by its signature deficits as by those features that are absent; aphasia, apraxia, agnosia, and visual field defect. Symptoms may be abrupt in onset or evolve over several hours, but in some instances the neurologic deficit evolves stepwise and relatively slowly, over as long a period as 2 to 3 days. Our experience has tended toward the shorter time frame, with most patients reporting that the full deficit is present not instantaneously but within minutes. Recovery, which may begin within hours, days, or weeks, is sometimes nearly complete even in the face of a severe initial stroke. However, many patients are left with some degree of clumsiness or slowness of movement of the affected side.

The motor disorder may take the form of a hemiparesis of the face and arm or arm and leg, or predominantly arm and proximal leg weakness; these fragmented patterns are indicative of a lesion located higher than the internal capsule, in the centrum semiovale. In these cases the stroke simulates an embolic stroke affecting the cortex.
A lacune of the lateral thalamus or (less often of the deep parietal white matter) is the cause of hemisensory defect involving the limbs, face, and trunk extending to the midline with no motor or language difficulty, a pure sensory stroke. Partial sensory syndromes involving only parts of the hemibody are less frequent than in motor lacunar strokes. The incidence, course, and outcome are much the same as in a pure hemiplegia.
As mentioned, in the ventral pons, the lacunar syndrome may be one of pure motor hemiplegia, mimicking that of internal capsular infarction except at times for relative sparing of the face and the presence of an ipsilateral paresis of conjugate gaze in some cases; or there is another highly characteristic lacunar syndrome of a combination of dysarthria and clumsiness of one hand. This “clumsy hand–dysarthria” stroke is usually located in the paramedian midpons on the side opposite the clumsy limb but a lacune in the posterior portion of the internal capsule on the side opposite the affected limb. Occasionally a lacunar infarction of the pons, midbrain, internal capsule, or parietal white matter gives rise to a hemiparesis with ataxia on the same side as the weakness (Fisher, 1965a; Sage and Lepore). Some of the brainstem syndromes may blend with basilar branch syndromes.
There are many other, less frequent, lacunar configurations but they can be identified by their similarity to one of the archetypal syndromes; they tend to affect one limited system or are fragments of a typical syndrome. Indeed, Fisher (1982) described 20 such variant types and several “miscellaneous” patterns. Some of these are difficult to accept, such as pure motor hemiparesis with confusion and loss of memory, but we have encountered many of the others, admittedly infrequently, including pure dysarthria, hemiballismus, virtual locked-in syndrome from bilateral lacunes in the base of the pons, and pure motor hemiplegia with sixth nerve palsy.
Some strokes that carry the term “lacune” are simply the result of larger deep cerebral infarctions along the lines of the striatocapsular stroke discussed earlier. In order to retain its clinical utility, the term lacune is probably best applied to small deep lesions that are the result of occlusion of a correspondingly small vessel and not to those strokes that result from occlusion of the orifices of several adjacent small vessels and are typically from larger atheromas in a parent vessel.
Multiple lacunar infarcts involving the corticospinal and corticobulbar tracts are a common cause of pseudobulbar palsy in clinical practice (trailed in frequency by amyotrophic lateral sclerosis and infiltrating tumors). Undoubtedly, an accumulation of lacunes deep in both hemispheres can give rise to gait disorders and also to mental dulling sometimes referred to as multiinfarct dementia (see further on and Chap. 21). The main differential diagnostic considerations are normal-pressure hydrocephalus (see Chap. 31) and the common degenerative brain conditions that affect the frontal lobes and basal ganglia (see Chap. 39).
Finally, it should be emphasized that what appears initially to be a lacunar syndrome may be the initial component or warning sign of a large deep territory infarction in the middle, posterior cerebral, or basilar arteries.
As mentioned earlier, MRI is more reliable than CT in demonstrating the lacunes. Initially, lacunes are seen on the MRI as deep oval or linear areas of T2, fluid-attenuated inversion recovery (FLAIR), and, especially, diffusion-weighted signal abnormality; later they become cavitated. Representative lacunar infarctions are shown in Fig. 34-16. The EEG, while little used for this purpose nowadays, may be helpful in a negative sense; in the case of lacunes in the pons or the internal capsule, there is a notable discrepancy between the unilateral paralysis or sensory loss and the negligible electrical changes over the affected hemisphere and, even with lacunes of the cerebrum, any EEG changes are disproportionately mild in comparison to the deficit.
Treatment of Ischemic Stroke
The main objective in these forms of ischemic cerebrovascular disease is the amelioration of the existing deficit and the prevention of future stroke. It is now a major goal of general medicine to reduce the incidence of stroke in the general population by the control of modifiable risk factors (“primary prevention”). In addition to reduction of known risk factors such as hypertension, smoking, and glucose control in diabetics, the widespread use of cholesterol-lowering statin medications has been shown in some studies to reduce the primary incidence of and recurrence of stroke. The treatment of stroke may be divided into three parts: management in the acute phase by measures to restore the circulation and arrest the pathologic process, physical therapy and rehabilitation, and measures to prevent further strokes and progression of vascular disease.
The relative advantages of placing the seriously ill acute stroke patient in a special “stroke” unit have been the subject of much study. The outcome in these patients in terms of morbidity and mortality is improved, although the differences have been small and difficult to document. However, this organizational plan has not been widely implemented and instead protocols for rapid evaluation of stroke and the emergence of a specialty of stroke neurology have proliferated instead. If nothing else, this is the result of a general recognition that stroke, like myocardial infarction, requires special expertise and focus. Protocols to prevent excessive hypertension after thrombolytic treatment are best instituted in units that have staffing patterns that create familiarity with these and other protocols. As already emphasized, the prevention of aspiration and pneumonia is paramount by identifying those patients at risk. The patients at risk also benefit from systematic application of protocols. Also deserving attention is the prevention of venous thrombosis in the legs, pulmonary embolism, and coronary syndromes. Some units find it advisable to keep patients supine for the first hours or day after an ischemic stroke, mainly to prevent hypotension and cerebral hypoperfusion; this approach has not been studied systematically. When sitting and walking begin, special attention should be given to the maintenance of normal blood pressure.
Several studies have confirmed the high prevalence of new or enhanced levels of hypertension immediately following an ischemic stroke and its tendency to decline over subsequent days even without medications. The treatment of previously unappreciated hypertension is preferably deferred until the neurologic deficit has stabilized. As suggested by Britton and colleagues, it is prudent to avoid antihypertensive drugs in the first few days unless there is active myocardial ischemia or the blood pressure is high enough to pose a risk to other organs, particularly the kidneys, or there is a special risk of cerebral hemorrhage as a result of the use of thrombolytic drugs.
Efforts are directed at establishing a diagnosis of thrombotic stroke at the earliest possible stage and circumventing the full deficit by all means available without risking the safety of the patient. Even when the symptoms and signs have become static, some of the affected tissue may not be irreversibly damaged and will survive if perfusion can be reestablished (the penumbra). If the patient is under care within 4.5 h of onset of the first symptom and this time can be established with confidence, thrombolytic therapy with tissue plasminogen activator (tPA) is usually indicated. The contraindications and implementation of thrombolytic therapy is given in detail in the next section.
Tissue plasminogen activators (recombinant tPA) convert plasminogen to plasmin. These drugs are effective in the treatment of coronary artery occlusion (but are associated with a 1 percent risk of cerebral hemorrhage) and have a reasonably clear role in the treatment of acute ischemic stroke. As an alternative to the usual form of tPA, alteplase and tenecteplase are genetically engineered mutant form of plasminogen activators. In the following discussion, we use “tPA” to represent all of the tissue plasminogen activators.
The benchmark study organized by the National Institute of Neurological and Communicative Disorders and Stroke (see the NINCDS and Stroke rtPA Stroke Study Group in the references) has provided evidence of benefit later from intravenous tPA when patients were examined months later. Treatment within 3 h of the onset of symptoms led to a 30 percent increase in the number of patients who remained with little or no neurologic deficit when reexamined 3 months after the stroke; this benefit persisted when assessed 1 year later in the study by Kwiatkowski and associates. It is not easy to comprehend why the benefits extended to all types of ischemic strokes, including those caused by occlusion of small vessels (lacunes) and why improvement was not apparent in the days immediately following treatment, only at later times.
The tPA in the NIH study was administered in a dose of 0.9 mg/kg, 10 percent of which was given as an initial bolus, followed by an infusion of the remainder over 1 h. This has been adopted as a conventional approach in practice. A dose of 90 mg was not exceeded, this being lower than the dose used for myocardial infarction. The relative improvement in neurologic state came at the expense of a 6 percent risk of symptomatic cerebral hemorrhage, i.e., a far lower rate than in most previous studies but twice the expected rate without thrombolysis (some of the hemorrhages were into the area of infarction and did not cause symptomatic worsening). Patients were excluded from the study if they had massive infarctions (encompassing more than two-thirds of the territory of the middle cerebral artery), had high scores on a clinical stroke scale that was devised for the National Institutes of Health (NIH) study (available at: and from other sources) had uncontrolled hypertension, were more than 80 years of age, or had recently received anticoagulants (except aspirin). Further analysis of the NINCDS trial revealed that patients who were treated earliest within the 3-h time frame had more benefit than those treated later; indeed, the administration of tPA in the time period between 2.5 and 3 h after the stroke was of less value.
Attempts to establish that patients with a longer duration of ischemic symptoms benefit from tPA have varied in success but have favored an effect up to 4.5 h as noted below. In particular, a subgroup of patients with carotid–middle cerebral artery strokes of moderate severity did appear to benefit. In some patients with basilar artery occlusion and coma of brief duration and those without extensive thrombosis, prompt tPA treatment also resulted in an overall improvement in neurologic function, but there were numerous exceptions. Following from these findings, a randomized trial of over 800 patients receiving tPA between 3 and 4.5 h after the onset of symptoms demonstrated that 10 percent more achieved a good outcome if given the drug than placebo and, although more had cerebral hemorrhages, the overall mortality was the same in the treated and untreated groups (Hacke et al, 2008). This study has altered practice to extend the acceptable interval for drug administration to 4.5 h if proper care is taken to select low-risk patients and prevent excessive blood pressure after tPA is given. A summary of the sequence of clinical trials that have led to the current recommendations for the use of tPA in acute stroke has been reviewed by Wechsler. Taken from a summary by the American Heart Association are the generally agreed upon inclusion and exclusion criteria for the use of intravenous tPA in Table 34-6. Intracranial and systemic bleeding is, of course, the great concern but a minor but interesting point is that some patients, who had been receiving angiotensin-converting enzyme inhibitors for the treatment of hypertension, seem to display angioneurotic edema as a side effect of tPA.
Inclusion criteria
|
Exclusion criteria
|
Relative contraindications
|
Generally excluded are those in whom the deficit is either very small (e.g., hand affected only, dysarthria alone, minor aphasia) or, more importantly, is so large as to implicate almost the entire territory of the middle cerebral artery. Many centers have expanded their practices beyond the confines of the NIH study, treating patients older than age 80 years and some with large strokes. Attempts are currently being made to identify patients in whom there is a mismatch between regional cerebral perfusion and preservation of brain tissue (typically shown by the absence of restricted water diffusion on MRI), but several studies so far have failed to demonstrate that selection of patients on this basis improves clinical outcome. Also ambiguous is the treatment of patients with acute stroke in whom the referable cerebral vessels are entirely patent.
An apparently effective ancillary technique has been to supplement intravenously administered tPA with 2 h of continuously applied transcranial ultrasound aimed at the occluded vessel. According to Alexandrov and colleagues, complete recanalization of the middle cerebral artery or clinical recovery occurred within 2 h but the outcome at 3 months did not differ from those who did not receive ultrasound in part because other similar trials were terminated prematurely as a result of high rates of cerebral hemorrhage. The technique has not been adopted into general clinical work. At least one small trial has shown alteplase to be slightly superior to tPA in restoring vascular patency if certain technical features of maintained cerebral perfusion were used to select patients (see Parsons and coworkers).
Although a valid approach to acute stroke, the use of acute thrombolytic therapy depends on the early identification of an as yet limited group of patients who arrive under care well before the 4.5 h from the onset; therefore this therapy is applicable to only a proportion of patients who present to the emergency department soon after the first symptoms (approximately 10 percent in most surveys) or those who have strokes while under observation in the hospital. It is noteworthy that attempts to reproduce the beneficial effects of tPA in a community setting have demonstrated deviations from treatment guidelines and an excess number of hemorrhages (Katzan et al). Nonetheless, acute intravenous thrombolysis that is managed by experienced individuals using validated protocols is an appropriate treatment for acute ischemic stroke. Public health education should increase the numbers of stroke patients who seek early attention and thus raise the proportion who are eligible for tPA treatment.
Thrombolytic substances injected intraarterially, or mechanical lysis for disruption or removal of an intravascular clot, can in some instances restore blood flow of the middle cerebral and basilar arteries. There is a high incidence of reocclusion of the treated vessel unless anticoagulants and stenting are also used. The most recent approaches involve one of a number of devices that retrieve clots from the intravascular lumen. Although early studies suggested that the results of intravascular clot lysis or removal were somewhat better than for intravenous or intraarterial thrombolysis, when more rigid criteria and trial design were applied, no such benefit has been shown.
In particular, three studies with different approaches to comparing these methods showed no clinical benefit for intraarterial means of restoring vascular patency in comparison to intravenous tPA (Broderick et al, and also Ciccone et al); and interventional methods were not superior when patients were selected on the basis of imaging patterns that demonstrated an area of ischemic tissue that was not yet infarcted (Kidwell and coworkers). Arguments notwithstanding that better mechanical devices will lead to improved outcomes, the results of systematic studies so far make it difficult to endorse intraarterial methods to revascularize cerebral vessels after acute stroke outside the confines of a clinical trial. The question of reversing the neurologic deficit from basilar artery thrombosis without cerebellar infarctions is nonetheless being studied.
This refers to the opening of the carotid artery or an area of intracranial atherosclerosis immediately after a stroke with the intention of improving the clinical outcome; the issue of endarterectomy for the prevention of future strokes is another matter and is taken up in a later section. In past decades, there had been limited experience with immediate surgical removal of a clot from the carotid artery or the performance of a bypass to restore function. Ojemann and colleagues (1995) operated on 55 such patients as an emergency procedure; 26 of these had stenotic vessels and 29 acutely thrombosed vessels. Of the latter, circulation was restored in 21, with an excellent or good clinical result in 16. In 26 patients with stenotic carotid arteries, an excellent or good result was obtained in 19. Usually several hours will have elapsed before the diagnosis is established. If the interval is longer than 12 h, opening the occluded vessel is usually of little value and may present additional dangers. In any case, this approach has been largely supplanted by the above-described endovascular techniques. Reoperation because the vessel has closed or caused an embolus immediately after carotid endarterectomy is a special circumstance in which rapid removal of a clot or repair of an intimal tear is performed more or less routinely and is also mentioned in a later section. This subject is reviewed again further on.
The separate issue of the endovascular treatment of intracranial atherosclerosis is considered here for convenience. The risks of manipulating intracranial vessels are obvious, particularly those of the circle of Willis with no surrounding tissue because they are located within the subarachnoid space. In an attempt to determine if a stent and angioplasty would improve outcome in patients who had TIAs or minor strokes as a result of an intracranial stenosis, Chimowitz and colleagues (2011) reported that their trial was stopped early because of poor outcomes of the group who were treated by stent in comparison to medical management. The treatment of symptomatic intracranial atherosclerosis therefore remains problematic and is delegated to the antiplatelet drugs and lipid-lowering agents discussed further on.
Several considerations weigh in any discussion of the institution and choice of antiplatelet or anticoagulant treatment (meant here to denote the agents that alter the clotting cascade) for stroke. First is the distinction between anticoagulation to prevent the progression of an acute stroke and the prophylactic use of anticoagulation for the prevention of future strokes. The pivotal issue in prevention of further strokes is whether the stroke or TIA is atherothrombotic or cardioembolic. As discussed further on, several studies point conclusively to a role for anticoagulation in stroke due to certain cardioembolic sources, particularly atrial fibrillation, while the indications in atherothrombotic disease are less certain.
Although not intuitive, if one holds the concept that a thrombus must form on an atherosclerotic plaque in order to cause stroke, it is apparent that anticoagulants have little effect at preventing the recurrence or progression of a thrombotic stroke and are certainly no more effective than agents that interfere with platelet aggregation. The two situations in which the immediate administration of heparin or an equivalent agent such as enoxaparin have drawn the most support from clinical practice are in basilar artery thrombosis with fluctuating deficits and in impending carotid artery occlusion from thrombosis or dissection. In these situations, the administration of heparin may be initiated while the nature of the illness is being clarified; the drug is then discontinued if contraindicated by new findings. Satisfactory clinical studies in support of this approach of acute anticoagulation have not been carried out and most authoritative writers find no evidence for the use of heparin in these situations (see, for example, Report of the Joint Stroke Guideline Development Committee authored by Coull et al). One fact seems fairly clear—that the administration of anticoagulants is not of great value once the stroke is fully developed.
Swanson has reviewed several trials evaluating heparin (including the International Stroke Trial and the TOAST study) and suggested that there was no net benefit from heparin in acute stroke because of an excess of cerebral hemorrhages. However, in these series there was a low incidence, estimated as 2 percent, of recurrent stroke in the first weeks after a cerebral infarction in the untreated groups. An early recurrent stroke rate this low almost precludes demonstrating a benefit from the use of heparin or heparinoid drugs. The issue of administering heparin in cases of recent cardioembolic cerebral infarction, particularly as a “bridge,” while waiting for the effects of warfarin to be established is addressed further on.
In the event heparin is used, and assuming tPA has not been used in the preceding 24 h, heparin may be given intravenously, beginning with a bolus of 100 U/kg followed by a continuous drip (1,000 U/h) and adjusted according to the partial thromboplastin time (PTT). Bleeding into any organ may occur when the PTT is greater than 3 times the pretreatment level. When the PTT exceeds 100 s, it is preferable to discontinue the infusion, check the blood clotting values, and then reinstitute the infusion at a lower rate based on the test results (rather than simply lower the infusion rate). In circumstances of fluctuating basilar artery ischemia, it is our practice to permit higher values of PTT.
The use of low-molecular-weight heparin (enoxaparin or nadroparin) given subcutaneously within the first 48 h of the onset of symptoms have uncertain benefit. In a limited trial, there was no increase in the frequency of hemorrhagic transformation of the ischemic region when compared to placebo treatment (Kay et al). Because the outcome measures in this study were coarse (death or dependence 6 months after stroke), further investigations of this approach need to be carried out. We can only infer that the use of low-molecular-weight heparin (approximately 4,000 U subcutaneously, tid) appears to be safe but there is no compelling evidence supporting their use in acute ischemic stroke.
The most convincing evidence favoring the efficacy of anticoagulants in the prevention of embolism comes from the Boston Area Anticoagulant Trial for Atrial Fibrillation. A group of patients at risk for stroke from chronic atrial fibrillation was randomized to be maintained for 2 years on warfarin (INR of 1.5 to 2) or no anticoagulation; there were 212 anticoagulated patients and 208 controls. Recurrent strokes were reduced by 86 percent in the warfarin group and the death rate was lower. One fatal hemorrhage occurred in each group; minor hemorrhages occurred in 38 of the warfarin-treated group and in 21 of the control group. In a similar study from Copenhagen, the incidence of stroke in a group receiving warfarin was calculated to be 2 percent per year in comparison to 5.5 percent per year in an untreated group. Several subsequent trials have attested to the efficacy of warfarin in the prevention of stroke in patients with nonrheumatic atrial fibrillation (see Singer). Subsequent studies suggest that an INR between 2 and 3 confers better protection than levels below 2. It should be pointed out, however, that patients younger than 65 years of age in these trials did not clearly benefit from long-term prophylactic anticoagulation unless there were additional cerebrovascular risk factors such as diabetes, hypertension, congestive heart failure, or cardiac valvular disease. Those younger than 65 years old and without such additional features (lone fibrillators), constituting about one-third of adults with atrial fibrillation, have a low risk of stroke. These observations are incorporated into the CHADS scores summarized in Table 34-3. Aspirin does not appear to afford the same degree of protective benefit as does anticoagulation, but some studies suggest a slightly better outcome than with no treatment; it has been used in the younger group of patients and in those unable to take warfarin.
Intracranial hemorrhage must be excluded by CT or MRI. A determination of prothrombin and partial thromboplastin activity is needed before therapy is started, but if this is not feasible, the initial doses of anticoagulant drugs can usually be given safely if there is no clinical evidence of bleeding anywhere in the body and there has been no recent surgery. Warfarin therapy, beginning with a dose of 5 to 10 mg daily, is relatively safe provided that the international normalized ratio (INR) is brought to 2 to 3 (formerly measured as a prothrombin time between 16 and 19 s) and the level is determined regularly (an approximate plan is once a day for the first 5 days, then 2 or 3 times a week for 1 to 2 weeks, and finally once every several weeks). Because there is no reliable evidence that complications are more frequent in the presence of mild to moderate hypertension if the INR is not allowed to exceed 2 to 3 times normal, we have not withheld anticoagulant therapy in these patients. However, when the blood pressure is greater than 220/120 mm Hg, an attempt is made to lower it gradually at the same time. Numerous drugs may alter the anticoagulant effects of the coumarins or add to the risk of bleeding—aspirin, cholestyramine, alcohol, carbamazepine, cephalosporin and quinolone antibiotics, sulfa drugs, and high-dosage penicillin being the most important ones. Hemorrhagic skin necrosis is a rare but dangerous complication. It is the result of a paradoxical microthrombosis of skin vessels and is liable to occur in patients with unsuspected deficiencies of endogenous clotting proteins (S and C). Although the disseminated form of skin necrosis occurs within days of initiating warfarin therapy, we have seen one patient with a form of this lesion following local skin injury after months on treatment.
For patients with atrial fibrillation of recent onset, an attempt should be made to restore normal sinus rhythm by the use of electrical cardioversion or a trial of antiarrhythmic drugs. If these fail, prophylactic anticoagulant therapy is recommended. Before attempting cardioversion of more long-standing atrial fibrillation anticoagulation for several days or longer is advisable to reduce emboli.
As noted below, opinions vary about the use and precise timing of instituting anticoagulation with warfarin after an embolic stroke, in part because the risk of recurrent stroke in the first days and weeks is low. In the past, there had been a theoretical risk of transient hypercoagulability when starting warfarin (on the basis of upregulation of protein S); this, and a desire to institute anticoagulation rapidly, led to a strategy of “bridging” with heparin or a low-molecular-weight heparin while awaiting the effects of warfarin to be evident. This risk has seemingly not had clinical significance. Therefore, on the basis of the aforementioned trials of acute anticoagulation that showed only a 1 to 2 percent frequency of early recurrent stroke, most clinicians have eschewed the “bridging” approach. The view that opposes heparin or a similar anticoagulant is based also on a retrospective study by Hallevi and colleagues, supported by a metaanalysis reported by Whiteley and coworkers, that found higher rates of symptomatic and serious bleeding in the brain or systemically with the bridging strategy.
In patients with very large cerebral infarcts that have a component of deep (basal ganglionic) tissue damage, especially in those patients who are also hypertensive, there may be a risk of anticoagulant-related hemorrhage into the acute infarct—”hemorrhagic conversion” (Shields et al). With the exception of urgent circumstances such as a mechanical heart valve that requires continuous anticoagulation, current practice has been to delay instituting warfarin when the stroke is large, a definition that is admittedly difficult to quantify—perhaps greater than half or a third of the territory of the middle cerebral artery supply. In these patients, anticoagulation therapy perhaps should be delayed for several weeks but even this is uncertain.
A frequent clinical problem arises in an elderly patient with atrial fibrillation who is at risk of falling from any of a number of causes including the stroke itself. In a review of selected administrative database records, Gage and colleagues concluded that the overall risk of inducing cerebral hemorrhage in older patients with atrial fibrillation treated with warfarin was lower than the risk of recurrent stroke. In those patients who had hemorrhages while receiving warfarin, they were, however, more likely to be fatal. Of course, decisions about anticoagulation must be tailored to the conditions of the individual patient. Alternatives to warfarin in patients with atrial fibrillation have been explored, such as the thrombin inhibitor ximelagatran, which has the advantage of not requiring monitoring by blood tests (see the Stroke Prevention Using an Oral Thrombin Inhibitor in Atrial Fibrillation [SPORTIF] trial).
Anticoagulant therapy may also be desirable for at least several weeks in patients with acute myocardial infarction, especially if the left side of the heart is involved. No guidelines have been established in these circumstances; the new common use of one or more platelet aggregation inhibitor drugs after myocardial infarction may preclude the concurrent use of warfarin. In cerebral embolism associated with bacterial endocarditis, anticoagulant therapy should be used cautiously because of the danger of intracranial bleeding, and one proceeds instead with antibiotics. Generally, we have not anticoagulated these patients.
Warfarin for the Prevention of Further Strokes in the Weeks and Months after a First Stroke Not due to Atrial Fibrillation
Despite its established benefit in preventing embolic strokes from atrial fibrillation, whether warfarin is effective in preventing strokes in patients with TIAs or recent stroke that are not due to atrial fibrillation is a question that has never been answered satisfactorily. Decisions regarding this matter depend not only on the putative ability of warfarin to prevent stroke but on an assessment of the relative risk of causing cerebral or systemic hemorrhage. The problem that continues to plague all attempts to use long-term anticoagulants, as already noted in the discussion regarding heparin in the acute situation, is the risk of hemorrhage estimated by Whisnant and colleagues to be 5 percent overall and considerably higher in elderly patients who have been treated for more than 1 year.
There had been a notion that warfarin is of some value in the first 2 to 4 months following the onset of an ischemic stroke due to atherosclerotic disease. However, the results of controlled studies have indicated that there is no reason to favor warfarin in comparison to aspirin in cases of atherothrombotic stroke. This was amply shown in the Warfarin-Aspirin Recurrent Stroke Study (WARSS; not including cardioembolic stroke) reported by Mohr and colleagues (2001); over 2 years the recurrent stroke rate was about 16 percent in both groups, and, surprisingly, the rate of cerebral hemorrhage was similar (near 2 percent). Similarly, for the special case of TIA or stroke that is shown to be because of intracranial atherosclerosis, Chimowitz and colleagues in the Warfarin-Aspirin Symptomatic Intracranial Disease (WASID) trial have suggested that warfarin provided no benefit over aspirin in preventing subsequent cerebrovascular events but warfarin had more risk as commented below. The WASID trial exposed not so much the deficiency of warfarin in prevention of stroke as much as the difficulties in its use, as pointed out by Koroshetz.
In contrast to the situation with atherothrombotic disease, warfarin is still used by some clinicians for prevention of a second stroke in cardioembolic disease. This applies more specifically to instances of a demonstrated source for the embolus in the cardiac chambers, valves, or through a patent foramen ovale as well as atrial fibrillation discussed earlier. The larger number of “cryptogenic strokes,” many of which may nevertheless be cardioembolic, cannot be confidently treated with warfarin. Of course, the extent of the evaluation to establish that an embolic stroke may have its origin in the heart, determines the likelihood of delineating a stroke as cryptogenic. In particular, several studies have shown that prolonged heart rhythm monitoring (event monitors) for up to 30 days moderately increases the rate of detection of atrial fibrilation. This creates a conundrum for the clinician confronted with a patient with an otherwise unexplained embolic stroke. There is therefore some latitude in deciding on treatment to prevent stroke in the subsequent months: none, aspirin (see below), or warfarin, as older studies had suggested. The issue of delaying anticoagulation after a large ischemic stroke was discussed in the preceding section.
The justification for, and excess risk of hemorrhage, when combining an antiplatelet agent with warfarin, has not been quantified. There seems to be relatively small risk if low-dose (81 mg) aspirin is included in a regimen of warfarin for atrial fibrillation. Beyond this statement, little can be said with authority but our personal experience, certainly tainted by the weaknesses engendered by the availability heuristic, is that subdural hematomas abound in elderly patients, with or without falls, who are on one or more agents. Even the relatively rapid reversal of anticoagulation with vitamin K and one of the clotting factor preparations or fresh plasma do little more than allow for safer surgery to remove intracerebral clots. Certain conditions such as chronic renal failure may confer a greater risk of cerebral hemorrhage with either drug, but the stroke risk from atrial fibrillation is also increased in comparison to patients with normal renal function as described in the epidemiologic study by Olesen and colleagues.
The introduction of factor Xa inhibitors has offered an alternative to the vitamin K antagonist, warfarin, for reducing strokes in patients with atrial fibrillation. In one study by Granger and coworkers that introduced apixaban for this purpose, there were slightly fewer strokes and fewer cerebral hemorrhages than with warfarin when the intended goal with warfarin was to keep the INR between 2 and 3. Dabigatran and rivaroxaban confer similar reductions in stroke to apixaban and warfarin (c.f., Patel et al). These drugs have the advantage over warfarin of not requiring regular blood tests for the measurement of the INR and of having fewer drug interactions. However, although they have short half-lives compared to warfarin, they are still present for many hours after discontinuation and anticoagulation is not easily reversible should there be systemic or cerebral bleeding.
Any type of serious bleeding with warfarin, even if not a result of overdosage, justifies immediate administration of fresh-frozen plasma and large doses of vitamin K. An INR above 5 in a patient who must remain anticoagulated—for example, one with a prosthetic heart valve—may be corrected with small doses of vitamin K (0.5 to 2 mg), preferably given intravenously.
Related posts:
 Disorders of the Special Senses
Chapter 20. Delirium and Other Acute Confusional States
Chapter 24. Fatigue, Asthenia, Anxiety, and Depression
Disorders of the Special Senses
Chapter 20. Delirium and Other Acute Confusional States
Chapter 24. Fatigue, Asthenia, Anxiety, and Depression
 Chapter 28. Normal Development and Deviations in Development of the Nervous System
Chapter 50. The Myotonias, Periodic Paralyses, Cramps, Spasms, and States of Persistent Muscle Fiber Activity
Chapter 28. Normal Development and Deviations in Development of the Nervous System
Chapter 50. The Myotonias, Periodic Paralyses, Cramps, Spasms, and States of Persistent Muscle Fiber Activity
 Chapter 48. Diseases of Muscle
Chapter 48. Diseases of Muscle
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


