Developmental Diseases of the Nervous System: Introduction
This broad heading subsumes a large number of both genetically driven developmental malformations and diseases acquired during intrauterine or early neonatal periods of life. They number in the hundreds according to the tabulation of Dyken and Krawiecki although many, if not most, are rare. Taxonomically, they make up two broad categories. The first includes specific gene defects, either mutations, deletions, or duplications of parts of genes (copy number variation), or single nucleotide polymorphisms that give rise to developmental aberrations or delays. The second category comprises a variety of environmental and infectious agents acting at different times on the immature nervous system during embryonal, fetal, and perinatal periods of life.
General Principles
Several points should be noted regarding the frequency of malformations. Jones, in the Smith monograph, has pointed out that a single minor malformation, usually of no clinical significance, occurs in 14 percent of newborns. Two malformations appear in 0.8 percent of newborns, and in this group, a major defect is five times more frequent than in the normal population. Three or more malformations are found in 0.5 percent of newborns, and in this latter group, more than 90 percent have one or more major abnormalities that seriously interfere with viability or physical well-being. The figures for major congenital malformations compiled by Kalter and Warkany are comparable but somewhat higher. What is most important for the neurologist is the fact that the nervous system is involved in most of infants with major malformations. Indeed, approximately 40 percent of deaths during the first postnatal year are in some manner related to prenatal malformations of the central nervous system.
Certain principles are applicable to the entire group of malformations. First, the abnormality of the nervous system is frequently accompanied by an abnormality of some other structure or organ (eye, nose, cranium, spine, ear, and heart), which relates them chronologically to a certain period of embryogenesis. Conversely, the presence of these malformations of nonnervous tissues suggests that an associated abnormality of the nervous system is developmental in nature. For example, the conjunction of cardiac, limb, gut, and bladder abnormalities with a neurologic disorder indicates the time at which the insult takes place: cardiac abnormalities occur between the fifth and sixth week; extroversion of the bladder at less than 30 days; duodenal atresia, before 30 days; syndactyly, before 6 weeks; meningomyelocele, before 28 days; anencephaly, before 28 days; cleft lip, before 36 days; syndactyly, cyclopia, and holoprosencephaly, before 23 days. Each is discussed in this chapter. This principle is not inviolable; in certain maldevelopments of the brain that must have originated in the embryonal period, all other organs are normal. One can only assume that the brain was more vulnerable than any other organ to prenatal as well as natal influences. Perhaps this occurs because the nervous system, of all organ systems, requires the longest time for its development and maturation, during which it is susceptible to disease. Low birth weight and gestational age, indicative of premature birth, increase the risk of cognitive or sensory developmental delay, seizures, and cerebral palsy.
A maldevelopment of whatever cause should be present at birth and remain stable thereafter, that is, be nonprogressive, in contrast to the majority of metabolic diseases of infancy discussed in the preceding chapter. However, this principle requires qualification: The abnormality may have affected parts of the brain that have not matured at birth, so that an interval of time must elapse postnatally before the defect can express itself.
Many of the teratologic conditions that cause birth defects pass unrecognized because they end in spontaneous abortions. For example, defects caused by chromosomal abnormalities occur in approximately 0.6 percent of live births, but such defects are found in more than 5 percent of spontaneous abortuses at 5 to 12 weeks gestational age.
Regarding the genetic causes of malformations and developmental delay, much has been learned in the past decade but a picture of the genetic influences on these conditions is still incomplete. For half a century, whole chromosome karyotyping allowed the recognition of conditions such as Down syndrome and its association with triplication of the entire chromosome 21. As more refined techniques became available, such as high-resolution banding, subtle changes such as small deletions in chromosomal architecture became apparent, as occur in Angleman and Prader-Willi syndrome and fragile-X syndrome. What followed, by the use of linkage analysis and other genetic techniques, was the elucidation of single gene mutations that were obligatorily associated with malformations or developmental delays; most of the inherited metabolic disorders of infancy and childhood discussed in Chap. 37 are of this type but also certain gross malformations of the brain such as lissencephaly, discussed further on.
These were the forerunners of a completely different category of technical innovations, anchored by the original method of sequencing short lengths of part of a gene by the Sanger method and its derivatives. With the development of the polymerase chain reaction and automated methods, longer and longer sequences of genes could be studied. This has allowed large population studies, merged with statistical techniques that expose genes in affected individuals that may be responsible for disease and are not present in high frequency in the general populations. From there, the rapid evolution of technology that is able to analyze thousands of portions of DNA in parallel and compare numerous sequences to a reference library of DNA has revealed variants at the resolution of single base pairs, single nucleotide polymorphisms (SNPs). Most exploration of human disease has been, until recently, based on the “common disease–common variant” model, in which a disease is attributable to limited number variants that exist in more than 1 to 5 percent of a population. For example, five polymorphisms are each responsible for doubling or tripling the risk of macular degeneration. However, most of these variants are probably not themselves responsible for the disease.
These types of genetic changes do not appear to explain the majority of the various developmental diseases. A newer concept of duplication or deletion of portions of genes, “copy number variation” is emerging as possibly explanatory of some proportion of diseases such as autism as discussed further on. What is interesting about copy number variation is that they give rise to several phenotypes of similar disorder, quite unlike conventional mendelian mutations. This indicates that copy number variants, like SNPs, probably are not directly causative of a condition but instead modulate other functions that express proteins. This is the situation for many of the forms of developmental abnormalities such as generic cognitive developmental delay, autism, and certain psychiatric diseases.
A textbook on the principles of neurology cannot catalog all the hereditary and congenital developmental abnormalities that affect the nervous system. For such details, the interested reader should refer to several excellent monographs. Three that the authors consult are Brett’s Pediatric Neurology, Berg’s Principles of Child Neurology, and Lyon and Evrard’s Neuropediatrie. These are supplemented by special atlases of congenital malformations mentioned further on. In this chapter, we sketch only the major groups and discuss in detail a few of the more common entities. The classification in Table 38-1 adheres to a grouping in accordance with the main presenting abnormality. Represented here are the common problems that lead families to seek consultation with the pediatric neurologist: (1) structural defects of the cranium, spine, and limbs, and of eyes, nose, ears, jaws, and skin; (2) disturbed motor function, taking the form of retarded development or abnormal movements; (3) epilepsy; and (4) developmental delay—mental retardation. The following discussion focuses on each of these clinical states.
|
Neurologic Disorders Associated with Craniospinal Deformities
A majority of the disorders in this group is the result of a genetic error, including those with a specific chromosomal abnormality. One has only to walk through an institution for the developmentally delayed to appreciate the remarkable number and diversity of dysmorphisms that attend abnormalities of the nervous system. Smith, in the third edition of his monograph on the patterns of human malformations, listed 345 distinctive syndromes; in the fourth edition (edited by K.L. Jones [1988]), many new ones were added. Indeed, a normal-appearing and severely cognitvely impaired individual stands out in such a crowd and will frequently be found to have an inherited metabolic defect or birth injury.
The intimate relationship between the growth and development of the cranium and that of the brain is likely responsible for many of the associations in maldevelopment. In embryonic life the most rapidly growing parts of the neural tube induce unique changes in, and at the same time are influenced by, the overlying mesoderm (a process termed induction); hence abnormalities in the formation of skull, orbits, nose, and spine are regularly associated with anomalies of the brain and spinal cord. During early fetal life the cranial bones and vertebral arches enclose and protect the developing brain and spinal cord. Throughout the period of rapid brain growth, as pressure is exerted on the inner table of the skull, the latter accommodates to the increasing size of the brain. This adaptation is facilitated by the membranous fontanels, which remain open until maximal brain growth has been attained; only then do they ossify (close). In addition, stature is apparently controlled by the nervous system, as shown by the fact that a majority of mentally retarded individuals are also stunted physically to a varying degree. Thus disorders of craniovertebral development assume importance not merely because of the physical disfigurement but also because they often reflect an abnormality of the underlying brain and spinal cord, whereby they become the main diagnostic signs of the maldevelopment.
Certain alterations in size and shape of the head in the infant, child, or even the adult, always signify a pathologic process that affected the brain before birth or in early infancy. Because the size of the cranium reflects the size of the brain, the tape measure is one of the most useful tools in pediatric neurology—no examination in a neurologically affected child is complete without a measurement of the circumference of the head. Graphs of head circumference in males and females from birth to 18 years of age were compiled by Nellhaus and are commonly used by pediatricians. A newborn whose head circumference is below the third percentile for age and sex and whose fontanels are closed may be judged to have a developmental abnormality of the brain. A head that is normal in size at term but fails to keep pace with body length (microcephaly) reflects a later failure of growth and maturation of the cerebral hemispheres (microencephaly).
This can be caused by factors extrinsic to the brain tissue, such as hydrocephalus and hydrancephaly (as defined below), or excessive brain growth (megalo- or macroencephaly; Table 38-2). The hydrocephalic head is distinguished by several features: frontal protuberance, or bossing; a tendency for the eyes to turn down so that the sclerae are visible between the upper eyelids and iris (sunset sign); thinning of the scalp and prominence of scalp veins; separation of the cranial sutures; and a “cracked pot” sound on percussion of the skull. Infantile hydrocephalus usually comes to medical attention because of an expanding cranium that exceeds normal dimensions for age. The usual causes are type II Chiari malformation, hereditary aqueductal stenosis, and prenatal infections, for example, toxoplasmosis. These disorders are discussed further on.
Hydranencephaly, defined as hydrocephalus and destruction or failure of development of parts of the cerebrum, is often associated with enlargement of the skull. When the cranium is transilluminated with a strong flashlight in a darkened room, the fluid-filled region of the cranium glows like a jack-o’-lantern. It can be caused by cerebral infarction from intrauterine vascular occlusion or by diseases such as toxoplasmosis and cytomegalovirus (CMV) disease, which destroy parts of each cerebral hemisphere. The lack of brain tissue reduces resistance to intraventricular pressure, permitting great enlargement of both lateral ventricles; it is especially marked if there is an added hydrocephalic state because of interference with cerebrospinal fluid circulation. This type of destruction of the cerebral mantle in the embryonal period may lead to the formation of huge brain defects with apposition of ventricular and pial surfaces (porencephaly) and subsequent failure of development of that part of the brain. Yakovlev and Wadsworth referred to the localized failure of evagination as schizencephaly and postulated that it was the result of a focal developmental defect in the wall of the cerebral mantle. They based their interpretation on the finding of malformed cortex in the margins of the defect but this might indicate only that the lesion preceded neuronal migration. Levine and coworkers attributed it to a destructive, possibly ischemic, lesion occurring in the first few weeks of gestation, at a time when neuronal migration was incomplete. However, at least some forms have been traced to genetic defects as detailed further on.
The macrocephalic head (a large head with normal or only slightly enlarged ventricles) may be indicative of an advancing metabolic disease that enlarges the brain, as in Alexander disease, Canavan spongy degeneration of infancy, and later phases of Tay-Sachs disease, all of which are described in Chap. 37. Agenesis of the corpus callosum, a common congenital defect, may be associated with macrocephaly and varying degrees of mental impairment, optic defects, and seizures. In a series of 56 patients with agenesis of the corpus callosum, Taylor and David reported the presence of epilepsy in 32 and varying degrees of developmental delay in 28; only 9 had no recognizable neurologic defects. Also noted was a high incidence of psychiatric disturbances in these patients. In such cases, CT and MRI reveal the characteristic “bat-wing” deformity of the ventricles. There is also asynchrony of electrical activity of the two cerebral hemispheres on the electroencephalogram (EEG). In a few of these patients, an autosomal dominant inheritance was found (Lynn et al). Agenesis of the corpus callosum is also part of the Aicardi syndrome (see further on) and the Andermann syndrome, and it has been noted, without explanation, in some cases of nonketotic hyperglycinemia.
Subdural hematomas also enlarge the child’s head and cause bulging of the fontanels and separation of the sutures. The infant is usually irritable and listless, taking nourishment poorly. Infants and children with neurofibromatosis, osteogenesis imperfecta, and achondroplasia also have enlarged heads; in the last of these, some degree of hydrocephalus appears to be responsible. Ultrasonography, which can be performed in the prenatal and neonatal periods, is usually diagnostic in all these cranial enlargements. Also MRI and CT scanning will disclose the size of the ventricles and the presence of subdural blood or fluid (hygroma).
Apart from patients with these pathologic states, there are individuals whose heads and brains are enlarged but who are normal in all other respects. Many of them come from families with large heads. Schreier and colleagues, who traced this condition through three generations of several families, declared it to be an autosomal dominant trait. This group represented 20 percent of 557 children referred to a clinic because of cranial enlargement, according to Lorber and Priestley.
This term refers to a marked enlargement of one cerebral hemisphere as a result of a developmental abnormality. The cortical gray matter and sometimes the basal ganglia are greatly increased in volume and weight. The cerebellum, brainstem, and spinal cord retain their normal dimensions. The cranium may be misshapen or enlarged but is normal in size in some cases. Rarely, the face and body are enlarged on the side of the enlarged hemisphere. The cortex of the giant hemisphere is thick and disorganized. Neurons are in disarray and some are enlarged; in some places the natural lamination of the cortex is effaced. Nothing is known about causation, but clearly embryogenesis has been deranged at the stage of neuroblast formation.
Clinically, these individuals are cognitively delayed and some have epilepsy. A degree of hemiparesis may be present but severe hemispheral neurologic deficits are generally not reported. However, hemimegalencephaly has been discovered at autopsy in a few individuals who had no mental or neurologic deficits.
Some of the most startling cranial deformities are caused by premature closure of the cranial sutures (membranous junctions between bones of the skull). Such conditions are estimated to occur in 1 of every 1,000 births, with predominance in males (Lyon and Evrard). The growth of the cranium is inhibited in a direction perpendicular to the involved suture(s), creating a compensatory enlargement in other dimensions as allowed by the patent sutures. For example, when the lambdoid and coronal sutures are both affected, the thrust of the growing brain enlarges the head in a vertical direction (tower skull, or oxycephaly, also referred to as turricephaly and acrocephaly). The orbits are shallow, the eyes bulge, and skull films show islands of bone thinning (lückenschädel). When only the sagittal suture is involved, the head is long and narrow (scaphocephalic) and the closed suture projects, keel-like, in the midline. With premature closure of the coronal suture, the head is excessively wide and short (brachycephalic). The nervous system is usually normal in these restricted craniostenoses. If this condition is recognized before 3 months of age, the surgeon can make artificial sutures that may permit the shape of the head to become more normal (Shillito and Matson). Once brain growth has been completed, little can be done aside from complex reconstructive surgery. When several sutures (usually coronal and sagittal) are closed, so as to diminish the cranial capacity, intracranial pressure may increase, causing headache, vomiting, and papilledema. An operation is then needed to increase the capacity of the skull.
In acrocephalosyndactyly, or Apert syndrome, craniostenoses are combined with syndactyly (fused, or webbed, fingers or toes). There are often added complications: mental retardation, deafness, convulsions, and loss of sight secondary to papilledema. The so-called clover-shaped skull is the most severe and lethal of the craniostenoses because of the associated developmental anomalies of the brain (see further on).
Approximately one-quarter of affected children with craniostenoses will be found to have a single gene or chromosomal abnormality, most commonly in the FGFR3 gene.
When, for any reason, an infant lies with the head turned constantly to one side (because of a shortened sternomastoid muscle or hemianopia, for example), the occiput on that side, over time, becomes flattened, as does the opposite frontal bone. The other occipital and frontal bones bulge, so that the maximum length of the skull is not in the sagittal but in the diagonal plane. This condition is called plagiocephaly, or wry head. Craniostenosis of one-half of a coronal suture may also distort the skull in this way.
Neuroembryologic studies have identified several milestones of neuroblast formation, migration, cortical organization, neuron differentiation, and connectivity. Certain developmental anomalies can be traced to one of these stages of cytogenesis and histogenesis in the first trimester of gestation and to the growth and differentiation that take place in the second and third trimesters. During the first trimester, postmitotic neurons that will ultimately reside in the cortex arise in the ventricular zone adjacent to the ventricles. They migrate along the scaffold of radial glia to form the multilayered cortex. It is interesting that neurons moving up the scaffold must pass through neurons that are already in position in the cortex, leading to an “inside-out” lamination in which the most recently born and arrived neurons reside on the outermost surface of the forming cortex.
Originally there is an excess of neurons, many of which degenerate during development—a process properly called apoptosis. There are recorded instances in which the full complement of neuroblasts and neurons fails to be generated. In the extreme, the emergence of two separate cerebral hemispheres may not occur (holoprosencephaly), or the bihemispheric brain may remain small (microcephaly). In other described instances, a diminished number of neurons are less obvious than their failure to migrate to the cortical surface; they remain scattered through the mantle zone in sheets and heterotopic aggregates. One type of focal band-shaped subcortical heterotopia is termed “double cortex.” Polymicrogyria refers to an excessive number of abnormally small gyri. It is expressed by a syndrome of mental retardation, seizures, delayed speech, and motor abnormalities. The cortex may fail to become sulcated—that is, it is lissencephalic or may be defectively convoluted, forming microgyric and pachygyric (broad gyral) patterns. In yet other brains, neuronal migration is normal for the most part, but small groups of neurons in particular regions may lag or present in regional heterotopias (focal dysgeneses; Fig. 38-1). These migrational disorders, particularly heterotopias, are now being recognized more often by MRI and are found to have a functional significance in epilepsy but also possibly in such states as nonspecific developmental delay, and dyslexia. Finally, the cortex may be normally formed and structured but there is a failure of differentiation of intra- and intercortical and interhemispheral connections, the most obvious one being agenesis of the corpus callosum.
The timing of embryogenesis of the main visceral organs and of the coincident stages of neural tube closure were given in the introduction. With these elementary facts of neuroembryology in mind, the bases of the following clinical states are readily conceptualized: anencephaly, lissencephaly, holoprosencephaly, polymicrogyria and pachygyria, microcephaly, and special combinations of cranial and somatic abnormalities. Each is described below.
In regard to disorders of brain development, there are also special types of tumors that are the consequence of abnormal neuronal or glial development. These are variously termed gangliomas, or gangliocytomas, dysembryoplastic neuroepitheliomas (DNETs), and low-grade astrocytomas. Sometimes they become manifest in the first year of life or even before birth. Their relatively slow growth and benign character suggest that some of them represent hamartomas rather than true neoplasms (see Chap. 31). Among the ones we have encountered in adults is the Lhermitte-Duclos type of cerebellar gangliocytoma that is characterized by a “tigroid” appearance on MRI (see Fig. 31-15).
Because each phase of cerebral development is under genetic control, it comes as no surprise that aberrant development might have a genetic basis. A singular advance in this field has been the identification in recent years of large numbers of genetic defects that underlie disorders of neuronal migration. These mutations, and what are known of their effects on the developing nervous system, were reviewed extensively by Mochida and Walsh, by Kato and Dobyn, and by Barkovich and colleagues; a summary is given in Table 38-3. The reader will notice that several quite different mutations may give rise to the same type of maldevelopment and that any given gene can cause malformations of varying severity but in most cases, the affected gene is active at a stage in brain development that makes the nature of the malformation understandable.
DISEASE | GENE | GENE FUNCTION |
|---|---|---|
Lissencephaly | ||
Lissencephaly with cerebellar hypoplasia | RELN (reelin) | Extracellular matrix protein |
Lissencephaly (Miller-Dieker) or isolated lissencephaly | LIS1 | Microtubule regulator |
X-linked lissencephaly with hypogonadism (Partington syndrome) | ARX (aristaless) | Transcription factor |
Muscle-eye-brain disease | POMGNT1 | Glycosyltransferase |
Walker-Warburg | POMT1 | Glycosyltransferase |
Holoprosencephaly | SHH (sonic hedgehog) | Transcription factor |
Double cortex | ||
Double-cortex or X-linked lissencephaly | DCX (doublecortin) | Microtubule-associated protein |
Heterotopias | ||
Periventricular nodular heterotopia | FLNA (filamin A) | Actin-binding protein |
Tuberous sclerosis | TSC1 (hamartin) | Tumor suppressor |
Tuberous sclerosis | TSC2 (tuberin) | Tumor suppressor |
Fukuyama muscular dystrophy | FCMD (fukutin) | Possible glycosyltransferase |
Schizencephaly | ||
Schizencephaly | EMX2 | Transcription factor |
Microcephaly | ||
Microcephaly | MCPH1 (microcephalin) | ? DNA repair |
Microcephaly | MCPH5 ASPM | Mitotic/meiotic spindle |
It should at the same time be noted that metabolic disturbances may also give rise to malformations of cerebral development. For example, in their review of the inborn errors of metabolism those are linked to cerebral dysgeneses. Nissenkorn and colleagues point out that disorders such as Zellweger syndrome and disorders of fatty oxidation, phenylketonuria (PKU), hyperglycinemia, and pyruvate dehydrogenase deficiency cause aberrant neuronal migration and dysgenesis of the corpus callosum.
This is one of the most frequent and also most appalling congenital malformations of the brain. Its incidence is 0.1 to 0.7 per all 1,000 births and females predominate in ratios ranging between 3:1 and 7:1 in different series. The concordance rate is low, both in identical and fraternal twins, but the incidence of the malformation is, nonetheless, several times the expected rate if one child in the sibship has been affected. Anencephaly has also been more frequent in certain geographic areas, for example, Ireland, for which various explanations of population genetics or environmental exposure have been postulated.
Missing in cases of anencephaly are large portions of scalp, cranial bones, and brain, including both cerebral cortex and white matter. All that remains is a hemorrhagic nubbin of nerve, glial, and connective tissue. Brainstem, cerebellum, and spinal cord are present but often, they too are malformed, as are the heart and other organs (15 to 40 percent of cases). In anencephalics who survive for a few days (65 percent die in utero and almost 100 percent before the end of the first postnatal week), startle reactions may be observed, as well as movements of limbs, spontaneous respirations, pupillary light reactions, ocular movements, and corneal reflexes. In a few, avoidance reactions, crying, and feeding reflexes can be elicited, indicating that only the rudimentary brain structures are required for these functions.
This condition, or a related one, can be anticipated if the mother’s serum levels of alpha-fetoprotein and acetylcholinesterase are elevated—even more reliably anticipated if they are elevated in the amniotic fluid. Positive tests should lead to ultrasonograph imaging of the fetus. Hydramnios is common.
The causes of anencephaly are multiple and include chromosomal abnormalities, maternal hyperthermia, and, apparently, deficiencies of folate, zinc, and copper (see Medical Task Force on Anencephaly). Of these, there is fairly secure evidence that supplemental intake of folic acid during the first trimester of pregnancy (i.e., from the time of conception) reduces the incidence of anencephaly and of myelomeningocele. Additional comments on anencephaly are given further on in this chapter in the section on “Dysraphism, or Rachischisis” (lack of fusion of the neural tube).
Included under this heading are several forms of defects of cerebral sulcation. In the lissencephalies, cortical convolutions may be absent altogether and there is morphologic evidence of several types of neuroblast deficiency. Such cases are of particular interest to neonatologists because of their associated physical abnormalities. The degree of impairment of neurologic function seldom allows longevity, so that relatively few affected individuals are found in institutions for the developmentally delayed. Seizures, poor temperature regulation, failure to accept nourishment, and apneic attacks combine to shorten life.
The failures of sulcation vary in severity. Neurons may fail to form or to migrate along glial projections to reach the more superficial layers of the cortex (a condition called in the past, Bielschowsky type); or the cortex, meninges, and eyes may fail to differentiate normally except for the dentate gyrus and hippocampus (Walker-Warburg type); or there may be other more minor focal derangements of cortical migrations and laminations with heterotopias of neurons in the white matter.
In the complete lissencephalies, the lateral and third ventricles enlarge because of a lack of the normal quantities of surrounding cerebral tissue (i.e., the aforementioned hydranencephaly). The cerebellar cortex is also abnormal. In some lissencephalic brains, there is slight sulcation presenting as abnormally broad or narrow convolutions, with thick, poorly laminated cortex; these are called pachygyrias or microgyrias, respectively, but the fundamental migratory abnormality is basically the same. The cerebellum is also abnormal, usually showing hypoplasia or aplasia involving the vermis or neocerebellum.
In the severe defects, the cranium is small at birth. In one type, which is inherited as an autosomal recessive trait, there are subtle craniofacial features (short nose, small mandible, ear abnormalities) as well as congenital heart disease. In another group, there is an associated familial congenital muscular dystrophy, placing the case between the Fukuyama and Walker-Warburg syndromes (see “Congenital Muscular Dystrophy” in Chap. 48).
Alobar and lobar holoprosencephalies are other examples of sulcation defects with craniofacial abnormalities in which development has gone awry in the fifth and sixth weeks of gestation (see Volpe, 1995). In these subtypes, the two cerebral hemispheres, either totally or only in part, form as a single telencephalic mass. In nearly all cases the cerebral defect is reflected by a single eye (cyclopia) and the absence of the nose, imparting an astonishing and diagnostic appearance.
Most of the severe disorders are sporadic, and the infants seldom survive for long. In a few of the malformations, a congenital infection with CMV or rubella has been implicated (Hayward et al).
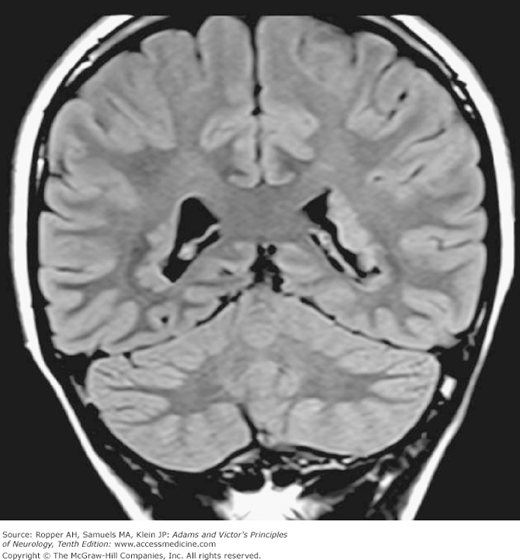
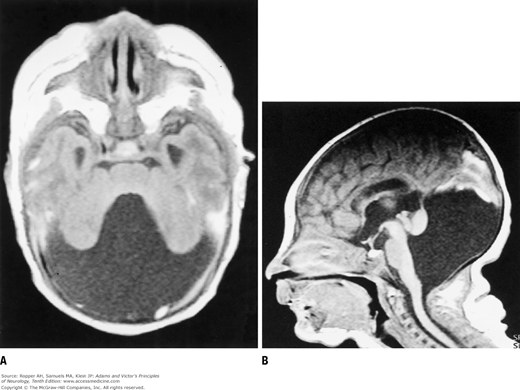
The Dandy-Walker syndrome represents a more restricted form of migration and neural tube defect. There is cerebellar vermian hypoplasia with or without hydrocephalus and, in some cases, an added agenesis of the corpus callosum with cerebral cortical dysgeneses (Landrieu). This defect, which is identified by the cystic enlargement of the fourth ventricle, is discussed further on, with the dysraphic neural tube defects.
That some instances of lissencephaly have a genetic basis has already been mentioned (see Table 38-3). Two genes that modify microtubular function have been identified: LIS1 and “doublecortin” or DCX. Large chromosomal deletions that span LIS1 cause Miller-Dieker syndrome, in which lissencephaly is associated with distinctive facial abnormalities; small defects in the same gene cause only lissencephaly. Lissencephaly with cerebellar hypoplasia is caused by mutations in the human “reelin” gene (RELN), the analogue of the defective gene in reelin mice (which have a reeling gait and abnormal cortical neuronal lamination). Defects in the transcription factor ARX are associated with X-linked lissencephaly, agenesis of the corpus callosum, and hypogonadism. Periventricular nodular heterotopia is caused by another gene defect, filamin A gene on the X chromosome. Some cases of holoprosencephaly have been traced to mutations in the sonic hedgehog gene.
In most of the above-described cerebral dysplasias, the cranium and brain are small, but there is also a primary form of hereditary microcephaly, called microcephaly vera, in which the head is astonishingly reduced in size (circumference less than 45 cm in adult life—i.e., 5 standard deviations below the mean). In contrast, the face is of normal size, the forehead is narrow and recedes sharply, and the occiput is flat. The brain often weighs less than 300 g (normal adult range: 1,100 to 1,500 g) and shows only a few primary and secondary sulci. The cerebral cortex is thick and unlaminated and grossly deficient in neurons. A few cases have an associated cerebellar hypoplasia or an infantile muscular atrophy. Stature is usually moderately reduced. Such individuals can be recognized at birth by their anthropoid appearance and later by their lumbering gait, extremely low intelligence, and lack of communicative speech. Vision, hearing, and cutaneous sensation are spared. In one of the cases studied by our colleagues, laborious effort using operant conditioning made it possible to teach the patient the shapes of simple figures. (His sister’s brain, examined by R.D. Adams, was malformed and weighed only 280 g.) Skull films show that the cranial sutures are present, as are convolutional markings on the inner table.
Lesser degrees of microencephaly have been associated with progressive motor neuron disease and degeneration of the substantia nigra (Halperin et al). Evrard and associates have described another rare type of microcephaly, which they call “radial microbrain.” The sulcal pattern is normal, and neuronal arrangements in the cerebral cortex are normal as well. The defect appears to be in the small number of neurons that are generated, not in their migration.
This category of maldevelopment is characterized by inappropriate migration of neurons to the pial surface, leading to a nodularity of the surface described as a cobblestone appearance. In the three disorders with this pathologic finding, the clinical picture is one of developmental delay conjoined with congenital muscular dystrophy. Three identified gene defects are thought to alter the glycosylation of critical proteins in the brain and in skeletal muscle. These genes include the gene fukutin in Fukuyama muscular dystrophy (see “Congenital Muscular Dystrophy” in Chap. 48), the POMGNT1 gene in muscle-eye-brain disease, and the POMT1 gene in Walker-Warburg syndrome, as summarized in Table 38-3.
There are so many cerebrosomatic anomalies that one can hardly retain visual images of them, much less recall all the physicians’ names by which they are known. There is great advantage in grouping these anomalies according to whether the extremities, face, eyes, ears, and skin are associated with a cerebral defect. The sheer number and variety of these anomalies permit only an enumeration of the more common ones and their most obvious physical characteristics. Unfortunately, apart from certain genetic linkages, no useful leads as to their origin have been forthcoming. Of necessity, one turns to atlases, one of the most thorough of which was compiled by Holmes and colleagues and is based on clinical material drawn in large part from the Fernald School and Eunice K. Shriver Center in Massachusetts. The reader may also turn to the texts by Gorlin and colleagues and by Jones for specific information. The older Ford’s Diseases of the Nervous System in Infancy, Childhood, and Adolescence is still a valuable reference, as is Jablonski’s Dictionary of Syndromes and Eponymic Diseases.
Fusion of two fingers or two toes or the presence of a tab of skin representing an extra digit may be seen at birth in an otherwise normal individual. However, when syndactylism is more severe and is accompanied by premature closure of cranial sutures, the nervous system usually proves to be abnormal as well. The general term acrocephalosyndactyly is used to describe the several combinations of craniostenotic and facial deformities and fusion of digits. Several of these disorders are a consequence of mutations in genes encoding one of two fibroblast growth factors or proteins related to them. The following descriptions include only the major features; most have, in addition, distinctive malformations of the orbits, ears, and palate.
Acrocephalosyndactyly types I and II (typical and atypical Apert syndrome). Turribrachycephalic skull, syndactyly of hands and feet (“mitten hands,” “sock feet”), moderate to severe mental retardation.
Acrocephalosyndactyly III (Saethre-Chotzen syndrome). Various types of craniostenoses, proximally fused and shortened digits, moderate degree of mental retardation. Transmission as an autosomal dominant trait.
Acrocephalosyndactyly IV (Pfeiffer syndrome). Turribrachycephaly; broad, enlarged thumbs and great toes; partially flexed elbows (radiohumeral or radioulnar synostoses); mild and variable mental retardation; autosomal dominant inheritance.
Acrocephalopolysyndactyly V (Carpenter syndrome). Premature fusion of all cranial sutures with acrocephaly, flat bridge of nose, medial canthi displaced laterally, excess digits and syndactyly, subnormal intelligence.
Acrocephalosyndactyly with absent digits. High, bitemporally flattened head; absent toes and syndactylic fingers; moderate mental retardation.
Acrocephaly with cleft lip and palate, radial aplasia, and absent digits. Microbrachycephaly because of craniostenosis, cleft lip and palate, absent radial bones, severe mental retardation.
Dyschondroplasia, facial anomalies, and polysyndactyly. Keel-shaped skull and ridge through center of forehead (metopic suture), short arms and legs, postaxial polydactyly and short digits, moderate mental retardation.
In all the foregoing types of syndactylism and cranial abnormalities, which may be regarded as variants of a common syndrome, the diagnosis can be made at a glance because of the deformed head, protuberant eyes, and abnormal hands and feet. The degree of cognitive delay proves to be variable, usually moderate to severe, but occasionally intelligence is normal or nearly so. The brain has been examined in only a few instances and not in a fashion to display fully the type and extent of this developmental abnormality.
Members of this group have distinctive anomalies of the cranium, face, and other parts, but craniostenosis is not a consistent feature.
Craniofacial dysostosis (Crouzon syndrome). Variable degrees of craniosynostosis; broad forehead with prominence in the region of the anterior fontanel region; shallow orbits with proptosis; midline facial hypoplasia and short upper lip; malformed auditory canals and ears; high, narrow palate; moderate mental retardation. As noted above, a genetic defect in one of the fibroblast growth factor receptors is responsible for about one-third of cases that are not associated with other deformities (Moloney et al). Autosomal dominant inheritance is seen in most cases.
Median cleft facial syndrome (frontonasal dysplasia; hypertelorism of Greig). Widely spaced eyes, broad nasal root, cleft nose and premaxilla, V-shaped frontal hairline, heterotypic anterior frontal fontanel (midline cranial defect); mild to severe mental retardation.
Chondrodystrophia calcificans congenita (chondrodysplasia punctata, Conradi-Hünermann syndrome). Prominent forehead; flat nose; widely separated eyes; short neck and trunk with kyphoscoliosis; dry, scaly, atrophic skin; cicatricial alopecia; irregularly deformed vertebral bodies; mental retardation infrequent. Severe shortening of limbs is seen in some cases.
Orofaciodigital syndrome. All the patients are female; they have pseudoclefts involving the mandible, tongue, maxilla, and palate; hypertrophied buccal frenula; hamartomas of tongue; sparse scalp hair; subnormal intelligence in one-half of cases.
Pyknodysostosis. Large head and frontal-occipital bossing, underdeveloped facial bones, micrognathia, unerupted and deformed teeth, dense and defective long bones with shortened limbs, short and broad terminal digits of fingers and toes, mental retardation in one-quarter of the cases.
Craniotubular bone dysplasias and hyperostoses. Included under this title are several different genetic disorders of bone, characterized by modeling errors of tubular and cranial bones. Frontal and occipital hyperostosis, overgrowth of facial bones, and widening of long bones occur in various combinations. Hypertelorism, broad nasal root, nasal obstruction, seizures, visual failure, deafness, prognathism, and retardation of growth are the major features.
In this category of anomalies, there is simultaneous failure or imperfect development of eye and brain. One member of this group, the oculocerebrorenal syndrome of Lowe, has already been mentioned and, of course, many of the mucopolysaccharidoses are characterized by corneal opacities, skeletal changes, and psychomotor regression as considered separately in Chap. 37. Also, congenital syphilis, rubella, toxoplasmosis, and CMV inclusion disease may affect retina and brain; hypoxia at birth requiring treatment with oxygen may injure the brain and lead to retrolental fibrodysplasia. The true developmental defects in this group are as follows:
Anophthalmia with mental retardation. Sex-linked recessive. Absent eyes; orbits and maxillae remain underdeveloped, but adnexal tissues of eyes (lids) are intact; subnormal intelligence. Some cases of anophthalmia have been ascribed to genes encoding transcription factors that play a role in the development of the neuraxis (SOX2, RAX, RAX6).
Norrie disease. Also sex-linked recessive; some sight may be present at birth; later, eyes become shrunken and recessed (phthisis bulbi); some have short digits, outbursts of anger, hallucinations, and possibly regression of psychomotor function. A novel gene, norrin, on the X chromosome has been implicated.
Oculocerebral syndrome with hypopigmentation. Autosomal recessive with absence of pigment of hair and skin; small, cloudy, vascularized corneas and small globes (microphthalmia); marked mental retardation; athetotic movements of limbs.
Microphthalmia with corneal opacities, eccentric pupils, spasticity, and severe mental retardation.
Aicardi syndrome with ocular abnormality. Chorioretinopathy, retinal lacunae, staphyloma, coloboma of optic nerve, microphthalmos, mental retardation, infantile spasms and other forms of epilepsy, agenesis of corpus callosum, and cortical heterotopias. The “batwing” deformity of the third and lateral ventricles on MR images and asynchronous burst-suppression discharges and sleep spindles are diagnostic. The condition is found only in females.
Lissencephaly of the Walker-Warburg type. This anomaly has already been mentioned, as has its association with congenital muscular dystrophy. Inheritance is autosomal recessive. Ocular lesions are a regular feature but of variable type (retinal dysplasia, microphthalmia, coloboma, cataracts, corneal opacities). There may be hydrocephalus, and CT scans and MRI disclose the lack of cerebral sulci (lissencephaly). The abnormal eyes and orbits and absence of cerebellar vermis are diagnostic (see Table 38-3).
Congenital tapetoretinal degeneration (Leber amaurosis). Visual loss from birth, moderate to severe mental retardation, and microcephaly. Early onset of blindness and absent electrical potentials on the electroretinogram (ERG) distinguish it from later-onset Leber optic atrophy, which is a mitochondrial disorder (see Chaps. 13 and 37).
Septooptic dysplasia (de Morsier syndrome). Diminished visual acuity, small optic discs, absence of septum pellucidum, and precocious puberty. Varying degrees of pituitary insufficiency may be present, requiring endocrine replacement.
These are less important from the neurologic standpoint, and mental retardation is present only in some cases.
Mandibulofacial dysostosis (Treacher-Collins syndrome, Franceschetti-Zwahlen-Klein syndrome)
Oculoauriculovertebral dysplasia (Goldenhar syndrome)
Oculomandibulodyscephaly with hypotrichosis (Hallermann-Streiff syndrome)
Midgets are abnormally small but perfectly formed people of normal intelligence; they differ from dwarfs, who are not only very small but whose bodily proportions are markedly abnormal and who may or may not be cognitively normal. It should be commented that a majority of severely cognitively delayed patients fall below average for height and weight, but there is a small group whose fully attained height is well below 135 cm (4.5 ft) and who stand apart by this quality alone (see Jones for Smith’s classification of dwarfs). The main types of dwarfism are as follows:
Nanocephalic dwarfism (Seckel bird-headed dwarfism). The uncomplimentary term bird head has been applied to individuals with a small head, large-appearing eyeballs, beaked nose, and underdeveloped chin. Such a physiognomy is not unique to any disease, but when combined with dwarfism it includes a few more or less specific syndromes. Up to 1976, approximately 25 cases had been reported, some with other skeletal and urogenital abnormalities, such as medial curvature of middle digits; occasional syndactyly of toes; dislocations of elbow, hip, and knee; premature closure of cranial sutures; and clubfoot deformity. These individuals are short at birth and remain so, living until adolescence or adulthood. Retardation is severe. The cause is a homozygous or compound heterozygous mutation in RAD3-realted protein, which is also implicated in ataxia-telangectasia. At autopsy the brain is found to have a simplified convolutional pattern; one of our patients had a type of myelin degeneration similar to that of Pelizaeus-Merzbacher disease.
Russell-Silver syndrome. Possibly an autosomal dominant pattern of inheritance, with short stature of prenatal onset, craniofacial dysostosis, short arms, congenital hemihypertrophy (arm and leg on one side larger and longer), pseudohydrocephalic head (normal-sized cranium with small facial bones), abnormalities of genital development in one-third of cases, delay in closure of fontanels and in epiphyseal maturation, elevation of urinary gonadotropins. Some cases appear to be caused by a nonmutational modification of genes, which are nonetheless inherited (imprinting).
Smith-Lemli-Opitz syndrome. Autosomal recessive inheritance with microcephaly, broad nasal tip and anteverted nares, wide-set eyes, epicanthal folds, ptosis, small chin, low-set ears, enlarged alveolar maxillary ridge, cutaneous syndactyly, hypospadias in boys, short stature, subnormal neonatal activity, and normal amino acids and serum immunoglobulins. Older survivors are bereft of language and are paraparetic, with increased reflexes and Babinski signs. The hips are usually dislocated. The responsible mutation is in DHCR7. The brain is small but has not been fully examined. Two of our patients are sibling girls.
Rubinstein-Taybi syndrome. Microcephaly but no craniostenosis, downward palpebral slant, heavy eyebrows, beaked nose with nasal septum extending below alae nasi, mild retrognathia, “grimacing smile,” strabismus, cataracts, obstruction of nasolacrimal canals, broad thumbs and toes, clinodactyly, overlapping digits, excessive hair growth, hypotonia, lax ligaments, stiff gait, seizures, hyperactive tendon reflexes, absence of corpus callosum, mental retardation, and short stature. This dominantly inherited disease is a result of disruption of so-called CREB-binding protein, a nuclear protein necessary for gene expression that is modulated by cyclic adenosine monophosphate (cAMP).
Pierre Robin syndrome. Possible autosomal recessive pattern of inheritance with microcephaly but no craniostenosis, small and symmetrically receded chin, glossoptosis (tongue falls back into pharynx), cleft palate, flat bridge of nose, low-set ears, mental deficiency, and congenital heart disease in half the cases. Camptomelia (bent bones) and diastrophic dwarfism (short limbs) are common.
DeLange syndrome (Cornelia DeLange syndrome). This phenotype shows some degree of variability but the essential diagnostic features are intrauterine growth retardation and stature falling below the third percentile at all ages, microbrachycephaly, generalized hirsutism and synophrys (eyebrows that meet across the midline), anteverted nostrils, long upper lip, and skeletal abnormalities (flexion of elbows, webbing of second and third toes, clinodactyly of fifth fingers, transverse palmar crease). All are moderately or, more often, severely retarded mentally, which, with the above craniofacial abnormalities, is diagnostic. It has been said, and it has been our experience, that many of these patients are prone to have a bad disposition, manifested by biting and spitting. Over half of cases are due to mutations in the NAPBL gene; the others involve SMC1A or SMC3.
Smith-Magenis syndrome. This is caused by deletions on chromosome 17, in which there is learning disability, severe behavioral problems (violence and self-injury), hyperactivity, deafness, and ocular abnormalities.
It is not surprising that skin and nervous system should share in pathologic states that impair development, as both have a common ectodermal derivation. Nevertheless, it is difficult to find a common theme in the diseases that affect both organs. In some instances, it is clear that ectoderm has been malformed from early intrauterine life; in others, a number of nondevelopmental acquired diseases of skin may have been superimposed. For reasons to be discussed later in this chapter, neurofibromatosis, tuberous sclerosis, and Sturge-Weber encephalofacial angiomatosis must be set apart in a special category of disease termed phakomatoses.
Hemangiomas of the skin are without doubt the most frequent cutaneous abnormalities present at birth, and usually they are entirely innocent. Many recede in the first months of life. However, an extensive vascular nevus located in the territory of the trigeminal nerve—and sometimes in other parts of the body as well—causes permanent disfigurement and usually portends an associated and topographically underlying cerebral lesion (Sturge-Weber syndrome).
Other neurocutaneous diseases are summarized below. A more complete review of these diseases will be found in the article by Short and Adams in Fitzpatrick’s Dermatology and the 1987 monograph by Gomez. The importance of recognizing the cutaneous abnormalities relates to the fact that the nervous system is usually abnormal, and often the skin lesion appears before the neurologic symptoms are detectable. Thus the skin lesion becomes a predictor of potential neurologic involvement.
Basal-cell nevus syndrome. This condition is transmitted as an autosomal dominant trait and is characterized by superficial pits in the palms and soles; multiple solid or cystic tumors over the head, face, and neck appearing in infancy or early childhood; mental retardation in some cases; frontoparietal bossing; hypertelorism; and kyphoscoliosis.
Congenital ichthyosis, hypogonadism, and mental retardation. This disorder is inherited as a sex-linked recessive trait. Aside from the characteristic triad of anomalies, there are no special features.
Xeroderma pigmentosum. The genetic pattern of inheritance is autosomal recessive. Skin lesions appear in infancy, taking the form of erythema, blistering, scaling, scarring, and pigmentation on exposure to sunlight; old lesions are telangiectatic and parchment-like, covered with fine scales; skin cancer may develop later; loss of eyelashes, dry bulbar conjunctivae; microcephaly, hypogonadism, and mental retardation (50 percent of cases). Kanda and associates classify this disease with what in the past had been called DeSanctis-Cacchione syndrome of “xerodermic idiocy” and believe the basic mechanism to be a faulty repair of DNA. They described two young adults with low intelligence, evidence of spinal cord degeneration, and peripheral neuropathy. The peripheral nerve lesions resembled those of amyloidosis, Riley-Day syndrome, and Fabry disease in that there was a predominant loss of small fibers. Other variants are described.
Sjögren-Larsson syndrome. Autosomal recessive with congenital ichthyosiform erythroderma, normal or thin scalp hair, sometimes defective dental enamel, pigmentary degeneration of retinae, spastic legs, and mental retardation.
Poikiloderma congenitale (Rothmund-Thompson syndrome). Autosomal recessive heredity; appearance of skin changes from the third to sixth months of life; diffuse pink coloration of cheeks spreading to ears and buttocks, later replaced by macular and reticular pattern of skin atrophy mixed with striae, telangiectasia, and pigmentation; sparse hair in half of the cases; cataracts; small genitalia; abnormal hands and feet; short stature; and mental retardation.
Linear sebaceous nevus syndrome. Here there is a linear nevus of one side of face and trunk, lipodermoids on bulbar conjunctivae, vascularization of corneas, mental retardation, focal seizures, and spike and slow waves in the EEG. Genetics remain uncertain.
Incontinentia pigmenti (Bloch-Sulzberger syndrome). Only females are affected; appearance of dermal lesions in first weeks of life; vesicles and bullae followed by hyperkeratoses and streaks of pigmentation, scarring of scalp, and alopecia; abnormalities of dentition; hemiparesis; quadriparesis; seizures; mental retardation; and up to 50 percent eosinophils in blood. The status of this disease is uncertain.
Focal dermal hypoplasia. Also a disease limited to females. Areas of dermal hypoplasia with protrusions of subcutaneous fat, hypo- and hyperpigmentation, scoliosis, syndactyly in a few, short stature, thin body habitus. Intelligence is occasionally subnormal.
Other rare entities are neurocutaneous melanosis, neuroectodermal melanolysosomal disease with mental retardation, progeria, Cockayne syndrome, and ataxia-telangiectasia (see Chap. 37; also Gomez, 1987).
Included under this heading is the large number of disorders of fusion of dorsal midline structures of the primitive neural tube, a process that takes place during the first 3 weeks of postconceptual life. Exogenous factors are presumed to be operative in most cases but there are genetic forms. The most extreme form is anencephaly, as described earlier; it is characterized by the absence of the entire cranium at birth, and the undeveloped brain lies in the base of the skull, a small vascular mass without recognizable nervous structures.
An eventration of brain tissue and its coverings through an unfused midline defect in the skull is called an encephalocele. Frontal encephaloceles may deform the forehead or remain occult. Associated defects of the frontal cortex, anterior corpus callosum, and optic-hypothalamic structures, as well as cerebrospinal fluid (CSF) leakage into frontal or ethmoid sinuses, pose a risk of meningitis. Some of these children are relatively normal mentally. Far more severe are the posterior encephaloceles, some of which are enormous and are attended by grave neurologic deficits such as blindness, ataxia, and mental retardation. However, lesser degrees of the defect are well known and may be small or hidden, such as a meningoencephalocele connected with the rest of the brain through a small opening in the skull. Small nasal encephaloceles may cause no neurologic signs, but if they are mistaken for nasal polyps and snipped off, CSF fistulae result.
A failure of development of the midline portion of the cerebellum referred to earlier, forms the basis of the Dandy-Walker syndrome (Fig. 38-2). A cyst-like structure, representing the greatly dilated fourth ventricle, expands in the midline, causing the occipital bone to bulge posteriorly and displace the tentorium and torcula upward. In addition, the cerebellar vermis is aplastic, the corpus callosum may be deficient or absent, and there is dilatation of the aqueduct as well as the third and lateral ventricles.
Even more frequent are abnormalities of closure of the vertebral arches. These take the form of a spina bifida occulta, meningocele, and meningomyelocele of the lumbosacral or other regions. In spina bifida occulta, the cord remains inside the canal and there is no external sac, although a subcutaneous lipoma or a dimple or wisp of hair on the overlying skin may mark the site of the lesion. In meningocele, there is a protrusion of only the dura and arachnoid through the defect in the vertebral laminae, forming a cystic swelling usually in the lumbosacral region; the cord remains in the canal, however. In meningomyelocele, which is 10 times as frequent as meningocele, the cord (more often the cauda equina) is extruded also and is closely applied to the fundus of the cystic swelling.
The incidence of spinal dysraphism (myeloschisis), like that of anencephaly, varies widely from one locale to another, and the disorder is more likely to occur in a second child if one child has already been affected (the incidence then rises from 1 per 1,000 to 40 to 50 per 1,000).
Exogenous factors (e.g., potato blight in Ireland) were many times implicated in an increased rate of both myeloschisis and anencephaly, but the effects of starvation and vitamin deficiency could never be separated from the potential effect of a toxic factor. It has now been established by numerous case-control and randomized treatment trials that inadequate intake of folate in early pregnancy is associated with an increased risk of these malformations. Folic acid, given before the 28th day of pregnancy is protective; vitamin A may also have slight protective benefit. Similar associations have been found with less certainty with exposure during pregnancy to certain antiepileptic drugs, particularly valproic acid and carbamazepine. Maternal diabetes and possibly obesity have been risk factors in some epidemiologic studies, as summarized by Mitchell and colleagues. The greatest risk, however, almost 30-fold higher, attaches to a previous pregnancy affected with spina bifida in particular.
As with anencephaly, the diagnosis can often be inferred from the presence of alpha-fetoprotein in the amniotic fluid (sampled at 15 to 16 weeks of pregnancy) and the deformity confirmed by ultrasonography in utero. Blood contamination is a source of error in the fetoprotein test (Milunsky). Acetylcholinesterase immunoassay, done on amniotic fluid, is another reliable means of confirming the presence of neural tube defects.
In the case of meningomyelocele, the child is born with a large externalized lumbosacral sac covered by delicate, weeping skin. The defect may have ruptured in utero or during birth, but more often the covering is intact. There is severe dysfunction of the cauda equina roots or conus medullaris contained in the sac. Stroking of the sac may elicit involuntary movements of the legs. As a rule the legs are motionless, urine dribbles, keeping the patient constantly wet, there is no response to pinprick over the lumbosacral dermatomal zones, and the tendon reflexes are absent. In contrast, craniocervical structures are normal unless a Chiari malformation is associated, as it often is (see further on). Differences are noted in the neurologic picture depending on the level of the lesion. If the lesion is entirely sacral, bladder and bowel sphincters are affected but legs escape; if lower lumbar and sacral, the buttocks, legs, and feet are more impaired than hip flexors and quadriceps; if upper lumbar, the feet and legs are sometimes spared and ankle reflexes retained, and there may be Babinski signs. The two common complications of these severe spinal defects are meningitis and progressive hydrocephalus from a Chiari malformation (see below). The subject of spina bifida and neural tube defects was reviewed by Botto and colleagues and by Mitchell and coworkers.
Prevention by the administration of folate during pregnancy is obviously paramount. Opinions as to proper management of the established lesion vary considerably. Excision and closure of the coverings of the meningomyelocele in the first few days of life are advised if the objective is to prevent fatal meningitis. After a few weeks or months, as hydrocephalus reveals its presence by a rapid increase in head size and enlargement of the ventricles, a ventriculoperitoneal shunt is required. Less than 30 percent of such patients survive beyond 1 year and the long-term results of treating these patients have not been encouraging. Lorber and colleagues report that 80 to 90 percent of their surviving patients are developmentally delayed to some degree and are paraplegic. The decision to undertake rather formidable surgical procedures is being questioned more frequently. Exceptionally, patients with meningomyelocele, and most of those with lumbar meningocele, are mentally normal.
The problems of meningomyelocele and its complications are so largely pediatric and surgical that the neurologist seldom becomes involved—except perhaps in the initial evaluation of the patient—in the treatment of meningeal infection, or in the case of shunt failure with decompensation of hydrocephalus. Of greater interest to the neurologist are a series of closely related abnormalities that produce symptoms for the first time in late childhood, adolescence, or even adult life. These include sinus tracts with recurrent meningeal infections, lumbosacral lipomas with low tethering of the spinal cord (“tethered cord”) causing an early childhood or delayed radicular or spinal cord syndrome; diastematomyelia, cysts, or tumors with spina bifida and a progressive myeloradiculopathy; and the Chiari malformation and syringomyelia that first present in adolescence or adult life. These abnormalities are described below.
Another class of disorders involves an occult lumbosacral dysraphism that is not inherited but is a result of faulty development of the cell mass that lies caudal to the posterior neuropore (normally this undergoes closure by the 28th day of embryonic life). Occult spinal dysraphism of this type is also associated with meningoceles, lipomas, and sacrococcygeal teratomas. Another well-recognized anomaly is agenesis of the sacrum and sometimes the lower lumbar vertebrae (caudal regression syndrome). Interestingly, in 15 percent of such cases, the mother is diabetic (Lyon and Evrard). Here there is flaccid paralysis of legs, often with arthrogrypotic contractures and urinary incontinence. Sensory loss is less prominent, mental function develops normally, and there is no hydrocephalus.
Another class of disorders involves an occult lumbosacral dysraphism that is not inherited but is a result of faulty development of the cell mass that lies caudal to the posterior neuropore (normally this undergoes closure by the 28th day of embryonic life). Occult spinal dysraphism of this type is also associated with meningoceles, lipomas, and sacrococcygeal teratomas. Another well-recognized anomaly is agenesis of the sacrum and sometimes the lower lumbar vertebrae (caudal regression syndrome). Interestingly, in 15 percent of such cases, the mother is diabetic (Lyon and Evrard). Here there is flaccid paralysis of legs, often with arthrogrypotic contractures and urinary incontinence. Sensory loss is less prominent, mental function develops normally, and there is no hydrocephalus.
Sinus tracts in the lumbosacral or occipital regions are of importance, for they may be a source of bacterial meningitis at any age. They are often betrayed by a small dimple in the skin or by a tuft of hair along the posterior surface of the body in the midline. (The pilonidal sinus should not be included in this group.) The sinus tract may lead to a terminal myelocystocele and be associated with dermoid cysts or fibrolipomas in the central part of the tract. Cloacal defects (no abdominal wall and no partition between bladder and rectum) may be combined with anterior meningoceles. Evidence of sinus tracts should be sought in instances of unexplained meningitis, especially when there has been recurrent infection or the cultured organism is of nosocomial dermal origin.
Of great interest are congenital cysts and tumors, particularly lipoma and dermoid, that arise in the filum terminale and attach (tether) the cord to the sacrum. Progressive symptoms and signs are produced as the spine elongates during development, thereby stretching the caudally fixed cord (Fig. 38-3). Some of these children have bladder and leg weakness soon after birth. Others deteriorate neurologically at a later age (generally between 2 and 16 years, sometimes later—see below). Complex disturbances of bladder function that produce urgency and incontinence beginning in the second or third decade may be the only manifestation, or the bladder symptoms may be combined with impotence (in the male) and numbness of the feet and legs or foot-drop (Pang and Wilberger). Several of our adult patients have had unusual visceral reflex reactions, such as involuntary defecation or priapism with stimulation of the abdomen or perineum. According to most surgeons, it is not the myelolipoma but the tethering of the cord that gives rise to symptoms; removal of the tumor is of little benefit unless the cord is detached from the sacrum at the same time. This may be difficult, for the lipoma may be fused with the dorsal surface of the spinal cord.
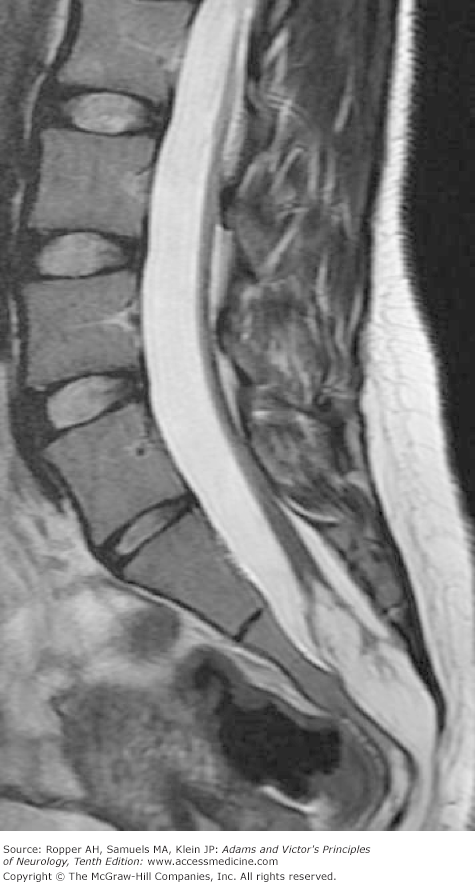
Diastematomyelia is another unusual abnormality of the spinal cord often associated with spina bifida. Here a bony spicule or fibrous band protrudes into the spinal canal from the body of one of the thoracic or upper lumbar vertebrae and divides the spinal cord into halves for a variable vertical extent. In extreme examples, the division of the cord may be complete, each half with its own dural sac and complete set of nerve roots. This longitudinal fissuring and doubling of the cord are spoken of as diplomyelia. With body growth, the restriction created by the bone spicule leads to a traction myelopathy, presenting with pain and progressive sensory, motor, and bladder symptoms, sometimes as late as adult life. Removal of the fibrous-bony spicule and untethering of the spinal cord have been beneficial in some cases.
Syringomyelia (see also Chap. 44) is a developmental cavity within the cervical cord, extending a variable distance caudally or rostrally, and usually associated with an Arnold-Chiari malformation that is described below. There are a variety of other neurodevelopmental spinal abnormalities in the high cervical region, such as fusion of atlas and occiput or of cervical vertebrae (Klippel-Feil syndrome), congenital dislocation of the odontoid process and atlas, platybasia, and basilar impression. These abnormalities are discussed in Chap. 44, with other diseases of the spinal cord.
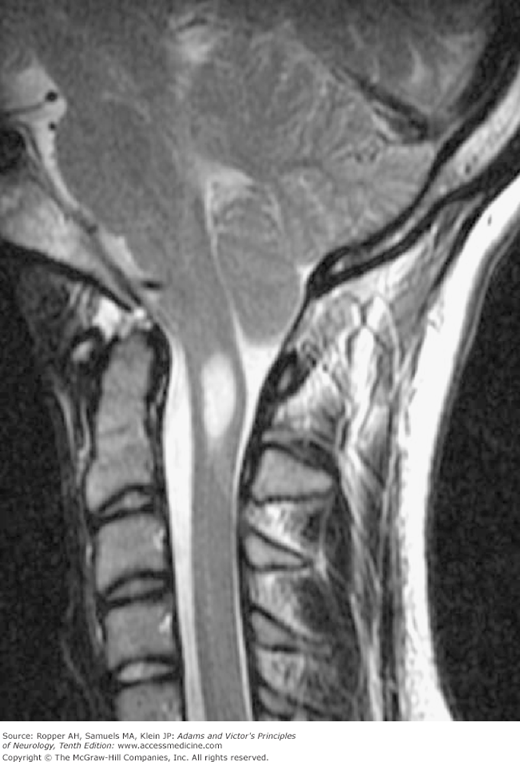
Encompassed by this term are a constellation of related congenital anomalies at the base of the brain, the most consistent of which are (1) extension of a tongue of cerebellar tissue posterior to the medulla and cord that extends into the cervical spinal canal; (2) caudal displacement of the medulla and the inferior part of the fourth ventricle into the cervical canal; and (3) a frequent but not invariable association with syringomyelia or a spinal developmental abnormality. These and associated anomalies were first clearly described by Chiari (1891). Several translations of his original material have been made, but they have been criticized as inaccurate. Arnold’s name is attached to the syndrome, but his contribution to our understanding of these malformations was relatively insignificant. Use of the double eponym Arnold-Chiari malformation is so entrenched that a dispute over its propriety serves little purpose. Chiari recognized four types of abnormalities. In recent years, the term has come to be restricted to Chiari’s types I and II—that is, to cerebellomedullary descent without and with a meningomyelocele, respectively. Type III Chiari malformation is no more than a high cervical or occipitocervical meningomyelocele with cerebellar herniation, and type IV consists only of cerebellar hypoplasia.
The incidence among adults, acquired from autopsy series and more recently, from incidentally discovered descent of the cerebellar tonsils on imaging procedures, is about 0.6 percent of the population. It should be emphasized that a proportion of normal individuals have a small tongue of the posterior cerebellum protruding by a few millimeters below the lower lip of the foramen magnum; this is usually of no significance and does not justify inclusion as a Chiari malformation. A historical account of the clinical, pathologic, and imaging aspects of this malformation and the evolution of ideas concerning it has been given by Bejjani.
Several other morphologic features are characteristic of the true Chiari anomaly. The medulla and pons are elongated and the aqueduct is narrowed. The displaced tissue (medulla and cerebellum) occludes the foramen magnum; the remainder of the cerebellum, which is small, is also displaced so as to obliterate the cisterna magna. The foramina of Luschka and Magendie often open into the cervical canal, and the arachnoidal tissue around the herniated brainstem and cerebellum is fibrotic. All these factors are probably operative in the production of hydrocephalus, which is always associated. Just below the herniated tail of cerebellar tissue there is a kink or spur in the spinal cord, which is pushed posteriorly by the lower end of the fourth ventricle. In this fully expressed form of the malformation, a meningomyelocele is nearly always found. It should again be emphasized that hydromyelia or syringomyelia of the cervical cord are commonly associated findings and the main theories of causation of the latter are based on the change in CSF dynamics produced by the Chiari malformation.
Developmental abnormalities of the cerebrum, particularly polymicrogyria may infrequently coexist, and the lower end of the spinal cord may extend as low as the sacrum (i.e., a tethered cord). There are usually cranial bony abnormalities as well. The posterior fossa is small; the foramen magnum is enlarged and grooved posteriorly. Nishikawa and colleagues suggested that smallness of the posterior fossa with overcrowding is the primary abnormality leading to the brain malformation. Often the base of the skull is flattened or infolded by the cervical spine (basilar impression).
In type II Chiari malformation (with meningomyelocele), the problem becomes one of progressive hydrocephalus. Cerebellar signs cannot be discerned in the first few months of life. However, lower cranial-nerve abnormalities—laryngeal stridor, fasciculations of the tongue, sternomastoid paralysis (causing head lag when the child is pulled from lying to sitting), facial weakness, deafness, bilateral abducens palsies—may be present in varying combinations. If the patient survives to later childhood or adolescence, one of the syndromes that are more typical of the type I malformation may become manifest.
In the more common type I Chiari malformation (without meningocele or other signs of spinal dysraphism), neurologic symptoms may not develop until adolescence or adult life. The symptoms are those of (1) increased intracranial pressure, mainly headache, (2) progressive cerebellar ataxia, (3) progressive spastic quadriparesis, (4) downbeating nystagmus, or (5) the syndrome of cervical syringomyelia (segmental amyotrophy and sensory loss in the hands and arms, with or without pain). Or the patient may show a combination of disorders of the lower cranial nerves, cerebellum, medulla, and spinal cord (sensory and motor tract disorders), usually in conjunction with headache that is mainly occipital. This combination of symptoms is easily mistaken for multiple sclerosis or a tumor at the foramen magnum. The symptoms are usually chronic but may have an acute onset after sustained or forceful extension of the neck, as, for example, after a long session of dental work, hairdressing in women, or chiropractic manipulation. The physical habitus of such patients may be normal, but approximately 25 percent have signs of an arrested hydrocephalus, or a short “bull neck.” When basilar impression (a congenital abnormality of the occipital bone that invaginates the posterior atlas into the cranial cavity) and a Chiari malformation coexist, it may be impossible to decide which of the two is responsible for the clinical findings.
The nature and severity of headache that are reasonably attributable to a Chiari malformation is somewhat unclear. Occipitonuchal pain with coughing, position change, or the Valsalva maneuver is the most dependable association, but even then, decompression may not relieve the symptoms. Exertional headache alone is a questionable association. Only large and genuine malformations, not minor descent of the tonsils should be considered causative. More generalized headaches may or may not be explained by the finding of a Chiari malformation and the advisability of a surgical treatment then depends on the degree of disability created by other aspects of the malformation. Further discussion of this subject is found in Chap. 10.
The tongue of cerebellar tissue and the kinked cervical cord obstruct the upward flow of CSF and give a highly characteristic imaging profile, particularly on sagittal MRI (Fig. 38-4). Inspection of the axial sections of scans at the level of the foramen magnum demonstrates crowding of the upper cervical canal by inferiorly displaced cerebellar tissue, but one must be aware of the variations in the normal position of the cerebellar tonsils at this level. A slight descent of the cerebellar tonsils that is reversible is also seen with low cerebrospinal fluid pressure and not indicative of a Chiari malformation. Recent inceptions in phase contrast MRI technology allow the imaging of CSF flow in the region of the foramen magnum but the relevance to choosing patients for surgical decompression has not been clarified (see summary by Menick). The CSF in Chiari malformation is usually normal but may show an elevated pressure and protein level in some cases for unexplained reasons.
The treatment of Chiari malformation and any associated basilar impression is far from satisfactory. If clinical progression is slight or uncertain, it is probably best to do nothing. If disability by way of spasticity, ataxia, pain in the shoulders or arms, or lower cranial-nerve disease is increasing, upper cervical laminectomy and enlargement of the foramen magnum are indicated. The proper course for patients that have headaches alone is uncertain but many such patients are operated upon if their cranial pain has been progressive, or if it is consistently and markedly worsened by cough or similar Valsalva actions, or if there is fainting or another associated symptom that can be reasonably related to the Chiari abnormality. The outcome of surgery, in our experience, has been less satisfactory when decompression was performed mainly for intractable headache, but there have been exceptions, especially when exertion or Valsalva maneuver elicits the symptoms.
The basic operation is suboccipital and C-1 decompression; various forms of shunting may be added if there is syringomyelia (the shunt is to the adjacent subarachnoid space) or hydrocephalus. The surgical procedure must be done cautiously. Opening of the dura and extensive manipulation of the malformation or excision of herniated cerebellum may aggravate the symptoms but is performed by some neurosurgeons to decompress the lower brainstem. Often, surgery halts the progress of the neurologic illness, arrests the hydrocephalus, or results in some other clinical improvement. The surgical series reported by Alzate and colleagues is representative. Emphasis was placed on proper patient selection, but the analysis of 66 cases, as in most other reports, was retrospective. (This reference is part of a very informative monograph). Most series report that a CSF leak follows surgery in about 5 percent of patients. The fate of an associated syringomyelia has been uncertain but most series report optimistic results.
As alluded to earlier, the role of phase-contrast MRI of CSF flow around the foramen magnum in selecting patients for decompression is unknown. The treatment of an associated syringomyelia and other developmental abnormalities in this region is discussed further in Chap. 44 under “Intramedullary Syringomyelic Syndrome.” We are unable to comment on the use by a limited number of neurosurgeons of posterior fossa decompression for the treatment of chronic fatigue syndrome and all manner of other symptoms except to say that it is entirely illogical, even when a Chiari malformation is detected.
Chromosomal Abnormalities (Karyotypic Chromosomal Dysgeneses)
As mentioned in the introduction, mid-twentieth-century discovery of outstanding significance was the recognition of a group of developmental anomalies of the brain and other organs associated with a demonstrable abnormality of the karyotype of autosomal and sex chromosomes. Jacobs and Lejeune in 1959 almost simultaneously were the first to note a triplication of the 21st chromosome in Down syndrome, and there followed the discovery of a number of other trisomies as well as deletions or translocations of other autosomal chromosomes and a lack or excess of one of the sex chromosomes. Such an event must take place sometime after the formation of the oocyte, during the long period when it lies fallow in the ovary or during the process of conception or germination and first cell divisions. Thus, all the cells in the embryo may carry the changed chromosome, or only some of them may carry it, the latter condition being called mosaicism.
The precise manner in which triplication or some other imperfection of a chromosome is able to derail the pathways of ontogenesis is a mystery. In some instances, a chromosomal imperfection may result from the lack of a gene or a distortion or fragmentation of an unstable gene, as in the fragile X syndrome. These germ line alterations are to be distinguished from acquired partial duplications and deletions of parts of genes that occur as acquired somatic mutations in many tremors and from variations in copy number of segments of genes that are emerging as possible explanations for a number of diseases such as autism.
Certain chromosomal abnormalities are incompatible with life, and it has been found that the cells of many abortuses and stillborns show abnormal karyotypes. Conversely, the organism may survive and exhibit any one of many syndromes, of which the following are the most frequent: (1) Down syndrome (mongolism, trisomy 21); (2) one type of arrhinencephaly (trisomy 13, Patau syndrome); (3) trisomy 18 (Edwards syndrome); (4) cri-du-chat syndrome (deletion of short arm of chromosome 5); (5) monosomy 21 (so-called antimongolism); (6) ring chromosomes; (7) Klinefelter syndrome (XXY); (8) Turner syndrome (XO); (9) others (XXXX, XXX, XYY, YY, XXYY); (10) fragile X syndrome, the most common form of inherited mental retardation; (11) Williams syndrome; and (12) Prader-Willi and Angelman syndromes. There are numerous less-frequent types, some of which are also discussed below because they have special neurologic interest. The overall frequency of chromosomal abnormalities in live births is 0.6 percent (see the review by Kalter and Warkany). For a comprehensive account of the chromosome-linked disorders the reader is referred to the article by Lemieux and for speculations on the nature of genetic retardations, can be found in the article by Nokelainen and Flint.
Described first in 1866 by Langdon Down, this is easily the best known of the chromosomal dysgeneses. Its frequency is 1 in 600 to 700 births, and it accounts for approximately 10 percent of all cases in every large series of cases of severe mental retardation. One cannot distinguish the Down syndrome with the common triplication of chromosome 21 from that caused by a translocation, and therefore duplication, of one arm, specifically the distal portion of the long arm that seems to contain the region responsible for the syndrome. Two genes have been of greatest interest in that region: DYRK1A and DSCR1.
Familiarity with the condition permits its recognition at birth, but the somatic appearance becomes more obvious with advancing age. The round head, open mouth, stubby hands, slanting palpebral fissures, and short stature impart an unmistakable appearance. The ears are low-set and oval, with small lobules. The palpebral fissures slant slightly upward and outward owing to the presence of medial epicanthal folds that partly cover the inner canthi (hence the old term mongolism). The bridge of the nose is poorly developed and the face is flattened (hypoplasia of the maxillae). The tongue is usually enlarged, heavily fissured, and protruded. Gray-white specks of depigmentation are seen in the irides (Brushfield spots). The little fingers are often short (hypoplastic middle phalanx) and incurved (clinodactyly). The fontanels are patent and slow to close. The hands are broad, with a single transverse (simian) palmar crease and other characteristic dermal markings. Lenticular opacities and congenital heart lesions (septal and other defects), as well as gastrointestinal abnormalities (stenosis of duodenum), are frequent. The patient with Down syndrome is slightly below average size at birth and is characteristically of short stature at later periods of life. The height attained in adult life seldom exceeds that of a 10-year-old child.
Hypotonia of the limbs is a prominent finding. At first, the Moro response is reduced or absent, and feeding is difficult. Most affected children do not walk until 3 to 4 years of age; their acquisition of speech is delayed, but over 90 percent talk by 5 years. The IQ is variable, and that of a large group follows a Gaussian curve with the median IQ being 40 to 50 and the range, 20 to 70. A placid, docile, and affectionate personality characterizes most Down patients. A high incidence of atlantoaxial instability puts these individuals at risk of traumatic spinal cord compression in athletic ventures.
An increased incidence of myelocytic and lymphocytic leukemia takes its toll. A number of patients have had embolic strokes and brain abscesses secondary to cardiac abnormalities and there is disproportionate occurrence of the rare cerebrovascular disorder known as moyamoya (see Chap. 34). Life expectancy is later shortened by the almost universal development of Alzheimer disease by the 40th year of life. This is explained by the presence on the duplicate copy of chromosome 21 of the gene for the precursor of the protein amyloid, a central factor in the development of Alzheimer disease. As Alzheimer disease develops, the usual clinical picture is marked by inattentiveness, reduced speech, impairment of visuospatial orientation, loss of memory and judgment, and seizures.
The triplication is found mostly in the offspring of older mothers, whereas the less-frequent translocation is found equally in the offspring of young and older women. In other subtypes of the Down syndrome, referred to as mosaics, some cells share in the chromosomal abnormality and others are normal. Affected individuals have atypical forms of the syndrome, and some such individuals are of normal intelligence.
The genetic changes that lead to cerebral maldevelopment and dysmorphic physical features are just beginning to be understood. A connection to genes that code for enzymes of folate has been suggested (among other mechanisms). The genetic aspects, as well as features pertaining to the medical care of these patients, are well summarized by Roizen and Patterson. They emphasize the high frequency of enteric sprue (celiac disease) and hypothyroidism and the need for screening for these conditions.
Brain weight is approximately 10 percent less than average. The convolutional pattern is rather simple. The frontal lobes are smaller than normal, and the superior temporal gyri are thin. There are claims of delayed myelination of cerebral white matter and also of immature and poorly differentiated cortical neurons. Alzheimer neurofibrillary changes and neuritic plaques are practically always found in Down patients who are older than 40 years of age, as mentioned.
It is possible to make the diagnosis of Down syndrome by demonstrating the chromosomal abnormalities in cells of the amniotic fluid. About one-third of pregnant mothers also have an abnormal elevation of serum alpha-fetoprotein in the second trimester of pregnancy. Other independent predictors of fetal Down syndrome are elevated serum chorionic gonadotropin and decreased estriol (Haddow et al). One can uncover a considerable proportion of the Down population by prenatal screening for these serum markers and by performing amniocentesis on women with positive tests to search for the chromosomal abnormality. Early detection is aided by the absence on imaging of a nasal bone between 11 and 14 weeks, at which time it is normally detected (Cicero et al).
These are listed here with brief descriptions of their main features.
Trisomy 13 (Patau syndrome). Frequency 1 in 2,000 live births, more female than male, average maternal age 31 years, microcephaly and sloping forehead, microphthalmos, coloboma of iris, corneal opacities, anosmia, low-set ears, cleft lip and palate, capillary hemangiomata, polydactyly, flexed fingers, posterior prominence of heels, dextrocardia, umbilical hernia, impaired hearing, hypertonia, severe mental retardation, death in early childhood.
Trisomy 18. Frequency 1 in 4,000 live births, more in females, average maternal age 34 years, slow growth, occasional seizures, severe mental retardation, hypertonia, ptosis and lid abnormalities, low-set ears, small mouth, mottled skin, clenched fists with index fingers overlapping the third finger, syndactyly, rocker-bottom feet, shortened big toe, ventricular septal defect, umbilical and inguinal hernias, short sternum, small pelvis, small mandible, death in early infancy.
Cri-du-chat syndrome (deletion in short arm of chromosome 5). Abnormal cry, like a kitten, severe mental retardation, hypertelorism, epicanthal folds, brachycephaly, moon face, antimongoloid slant of palpebral fissures, micrognathia, hypotonia, strabismus.
Ring chromosomes. Mental retardation with variable physical abnormalities.
Klinefelter syndrome (XXY). Only males affected. Eunuchoid appearance, wide arm span, sparse facial and body hair, high-pitched voice, gynecomastia, small testicles, usually developmentally delayed but not severely so; high incidence of psychosis, asthma, and diabetes.
Turner syndrome (XO). Only females affected. Triangular face, small chin, occasionally hypertelorism and epicanthal folds, widely spaced nipples, clinodactyly, cubitus valgus, hypoplastic nails, short stature, webbed neck, delayed sexual development, mild developmental delay. The manner of inheritance of the X chromosome may have bearing on the patient’s personality and level of functioning, as noted in Chap. 21.
Colpocephaly. A rare type of malformation of the brain consisting of marked dilatation of the occipital horns of the lateral ventricles, thickening of the overlying rim of cortical gray matter, and thinning of the white matter. The associated clinical picture comprises developmental delay, spasticity, seizures, and visual abnormalities (because of optic nerve hypoplasia). This disorder is probably of diverse causation, but it is listed here with the chromosomal abnormalities because some cases have been associated with the mosaicism for trisomy 8 (Herskowitz et al). The term colpocephaly is often used incorrectly to apply to all forms of ventricular enlargement (including hydrocephalus) associated with abnormal development of the brain.
Fragile X syndrome (see further on for additional clinical details in the section on developmental delay). This abnormality is among the most common inherited forms of developmental delay, estimated to occur in 1 of every 1,500 male live births and accounting for 10 percent of severe developmental delay in males. Females, with two X chromosomes, are affected about half as frequently, and then only to a slight degree. With the advent of new markers for the detailed structure of chromosomes, Lubs observed an unusual site of frequent breakage (“fragility”) on the X chromosome and related it to a syndrome that included developmental delay, flaring ears, elongated facies, slightly reduced cranial perimeter, normal stature, and enlarged testes. More recently, rare progressive ataxia has been reported in adults who harbor the chromosomal abnormality and had displayed little or no cognitive deficiency.
The chromosomal fragility appears to be due to a heritable, unstable CGG repeating sequence in the X chromosome. Affected individuals have over 230 repeats and carriers have 60 to 230 repeats. The prolonged sequence inactivates a gene (FMR1) that codes for an RNA-binding protein of as yet an unknown connection to brain function. The genetics of this and other retardations was reviewed by Nokelainen and Flint. Rousseau and colleagues described a simple and sensitive test using DNA analysis for the diagnosis of the syndrome both prenatally and after birth. Because of mosaicism, the length of the triplet repeat does not directly relate to the degree of expression of retardation, and the fragile X alteration occasionally turns up in mentally normal males; in some instances, the male children of their daughters have the disease. In some of the cases we have observed, the intellectual deficit has been mild in degree and the main abnormalities have taken the form of troublesome behavior, logorrhea, an autistic type of gaze aversion, and asociality. These and other neurobehavioral features of the syndrome and its unique pattern of inheritance (it is neither recessive nor dominant) have been discussed by Shapiro.
Williams syndrome. Described by J.C.P. Williams and colleagues of Australia from the perspective of a supravalvular aortic stenosis and a year later by Beuren and colleagues, this unique combination of cerebral maldevelopment and cardiovascular abnormalities has been traced in most patients to a microdeletion on chromosome 7 in the region of the gene that codes for the protein elastin. Its frequency is 1 in 20,000 newborns. Further discussion of clinical features of this syndrome is found later in the chapter.
Prader-Willi and Angelman syndromes. The Prader-Willi syndrome was already mentioned in relation to the hyperphagia of hypothalamic disorders (adiposogenital dystrophy, Froehlich syndrome). It is not uncommon (1 in 20,000 births) and affects both sexes equally. Hypotonia (floppy infant), areflexia, small stature, dysmorphic facies, and hypoplastic genitalia are evident, and arthrogryposis may be present at birth. After the first year, developmental delay becomes obvious and obesity, due to hyperphagia, becomes prominent. Patients are identified by the “H3O” mnemonic, referring to hypomentia, hypotonia, hypogonadism, and obesity. The disorder is associated with a deletion at 15q11-q13 (a so-called microdeletion, as in Williams syndrome), which can be identified by a combination of cytogenetic and DNA analyses. In 70 percent of cases the disease is caused by a noninherited deletion from the paternal X chromosome.
The Angelman syndrome, another cause of severe developmental delay, is associated with the identical chromosomal abnormality to that found in the Prader-Willi syndrome, but there is usually a maternally inherited single-gene defect. The difference in phenotype derives from a complex genetic phenomenon termed spatially restricted imprinting. The phenotype comprises severe developmental delay, microcephaly, refractory seizures, absence of speech, ataxia, inappropriate laughter, prominent jaw, thin upper lip, and prolonged tongue. Outstanding are an unusual marionette-like stance coupled with a persistent tendency to laugh and smile (hence the old name “happy puppet syndrome”; see also Chap. 37).
Rett syndrome, discussed more fully further on, is mentioned here because it is to the result of a dominant defect on the X chromosome. It affects 1 of every 10,000 to 15,000 girls. After 6 to 18 months of normal development, motor skills and mental abilities seem slowly to regress. Certain handwringing and other stereotyped hand movements appear as the disease progresses and are characteristic.
Several generalizations can be made about these chromosomal dysgeneses. First, the autosomal ones are often lethal (Rett syndrome is an exception), and they almost always have a devastating effect on cerebral growth and development, whether the infant survives or not. Anomalies of nonneural and a degree of externally visible dysmorphism structures are regularly present—an association so constant that one may safely predict that an otherwise normally formed infant will not have a detectable chromosomal defect. However, only in the Down syndrome and trisomy 13 (and possibly trisomy 18) are the physiognomy and bodily configuration highly characteristic. Surprisingly, some of the most grotesque disfigurements, such as anencephaly and multiple severe congenital anomalies, are not related to a morphologic abnormality of chromosomes. By contrast, an insufficiency of sex chromosomes induces only subtle effects on the brain, affecting intellect and personality; to some extent this is true of supernumerary sex chromosomes (XYY, for example).
The basic abnormality of the brain underlying the developmental delay in many of these chromosomal dysgeneses has not been ascertained. The cerebrum is slightly small, but only minor changes are seen in the convolutional pattern and cortical architecture in conventional microscopic preparations. Neurocellular methodologies to date are not sufficiently advanced to reveal the fundamental cerebral abnormality.
A number of observations have repudiated the former belief that the human embryo is naturally shielded against exogenous causes of maldevelopment. Irradiation during the first trimester, rubella and CMV infections, severe hypothyroidism of the mother during this same period, and the action of alcohol, vitamin A, and thalidomide have all been observed, among a multitude of other agents, to give rise to serious disorders of development. Quite relevant to the neurologist, the offspring of mothers receiving anticonvulsant drugs during the early months of pregnancy have a slightly increased risk of developing birth defects (approximately 5 percent, compared to 3 percent for the general population—see “Teratogenic Effects of Antiepileptic Medications” in Chap. 16). Cleft lip and palate are the most common anomalies attributable to anticonvulsant drugs; other craniofacial defects, spina bifida, minor cardiac defects, and dysraphisms have also been reported at a slightly increased rate. Claims and counterclaims have been made concerning the pathogenicity of numerous other substances. Mainly, the data are from animals given amounts far in excess of any possible therapeutic doses in humans. The data from humans are so meager from a multitude of such substances that they are not discussed here. The reader may refer to the article by Kalter and Warkany for further information.
The Phakomatoses (Congenital Neuroectodermoses)
As stated earlier, there are two broad categories of neurocutaneous diseases. In one, the infant is born with a special type of skin disease or develops it in the first weeks of life; in the other forms, the cutaneous abnormality, although often present in minor degree at birth, later evolves as quasineoplastic disorders. The quasineoplastic disorders to which van der Hoeve in 1920 applied the term phakomatoses (from the Greek phakos, meaning “mother spot,” “mole,” or “freckle”) are tuberous sclerosis, neurofibromatosis, and cutaneous angiomatosis with central nervous system (CNS) abnormalities, which have many features in common: hereditary transmission, involvement of organs of ectodermal origin (nervous system, eyeball, retina, and skin), slow evolution of lesions in childhood and adolescence, a tendency to form hamartomas (benign tumor-like formations because of maldevelopment), and a disposition to fatal malignant transformation. These disorders are discussed below and listed in Table 38-4.
True phakomatoses
|
Cutaneous angiomatosis with abnormalities of the central nervous system
|
Tuberous sclerosis is a congenital disease of hereditary type in which a variety of lesions, because of a limited hyperplasia of ectodermal and mesodermal cells, appear in the skin, nervous system, heart, kidney, and other organs. It is characterized by the triad of adenoma sebaceum, epilepsy, and developmental delay. Hypomelanotic skin macules (“ash-leaf” lesions) and the subepidermal fibrotic “shagreen patch” are diagnostic features.
It is stated that Virchow recognized scleromas of the cerebrum in the 1860s and that von Recklinghausen reported a similar lesion combined with multiple myomata of the heart in 1862, but Bourneville’s articles, appearing between 1880 and 1900, presented the first systematic accounts of the disease and it was he who related the cerebral lesions to those of the skin of the face. Vogt (1890) fully appreciated the significance of the neurocutaneous relationship and formally delineated the triad of facial adenoma sebaceum, epilepsy, and developmental delay. “Epiloia,” a term for the disease introduced by Sherlock in 1911, never gained general acceptance. These and other historical aspects are reviewed in Gomez’s monograph.
The disease has been described in all parts of the world and is equally frequent in all races and in both sexes. Heredity is self-evident in only a minority of cases— 50 percent in some series and as little as 14 percent in the series of Bundag and Evans (cited by Brett). The disease is determined by two autosomal dominant genes (see below), but it has been estimated as 1 in 20,000 to 300,000. The disease is inherited in an autosomal dominant fashion but with variable penetrance. The abnormal gene may be in one of two sites—the long arm of chromosome 9, designated as TSC 1 (hamartin), or in the short arm of chromosome 16, TSC 2 (tuberin), which is common. A wide variety of mutations have been described and both alleles must be affected for expression of the disease (“loss of heterozygosity”). Approximately 15 percent of sporadic cases show no identifiable mutation and tend to have milder manifestations, perhaps on the basis of mosaicism. Hamartin and tuberin function as tumor suppressor proteins and interact to suppress cell growth. This may, in part, explain the proclivity to develop various growths and hamartomas. The cerebral lesions and two of the three associated skin lesions of tuberous sclerosis are of this type. Several hypotheses relating to neuronal migration or to excessive secretion of growth factors have been proposed to link the inactivation of these genes with the pathogenesis of the characteristic lesions. Much of the work in understanding the function of these two proteins and their role in tumor formation has been performed in Drosophila and are summarized in the extensive review by Crino and colleagues.
The disease involves many organs in addition to the skin and brain and it may assume a diversity of forms, the least severe of which (i.e., the forme fruste) is difficult to diagnose; hence, one cannot be precise about its incidence. Tuberous sclerosis accounts for about 0.66 percent of the developmentally delayed in institutions and 0.32 percent of epileptics. The medical literature contains a number of reports of patients whose mentality is preserved and who have never had convulsions.
The cellular elements within the nodular cerebral lesions (called tubers; see below) are abnormal in number, size, and orientation. The tumor-like growths in different organs may include cells of more than one type (e.g., fibroblasts, cardiac myoblasts, angioblasts, glioblasts, and neuroblasts), and their number is locally excessive. Something appears to have gone awry with the proliferative process during embryologic development, yet it is kept under control, in the sense that only rarely do the growths undergo malignant transformation. Highly specialized cells within the lesions may attain giant size; neurons 3 to 4 times normal size may be observed in the cerebral scleroses. These facts emphasize the potentially blastomatous character of the process.
Surgically resected tubers show activation of a cell-size control pathway (mammalian target of rapamycin [mTOR]); this is in keeping with the effects of TSC mutations on this cascade and probably explains the giant neurons as mentioned further below.
Cutaneous and ectodermal |
Shagreen patch |
Facial angiofibromas (adenoma sebaceum) |
Ungular and subungual fibromas |
Hypomelanotic skin macules (more than 3) |
Multiple dental enamel pits |
Bone cysts |
Hamartomatous rectal polyps |
Gingival fibromas |
Retinal achromatic patch |
Multiple renal cysts |
Cardiac rhabdomyoma |
Renal angiomyolipoma |
Lymphangiomatosis |
Neural |
Seizures |
Developmental delay |
Cortical tubers |
Subependymal nodules |
Subependymal giant cell “astrocytoma” |
Retinal hamartoma |
The disease may be evident at the time of birth (the diagnosis has been made by CT scan in neonates), but more often the infant is judged at first to be normal. In approximately 75 percent of cases, attention is drawn to the disease initially by the occurrence of focal or generalized seizures or by slowed psychomotor development. As with any condition that leads to developmental delay, the first suspicion is raised by delay in reaching normal maturational milestones. Whatever the initial symptom, the convulsive disorder and developmental delay become more prominent within 2 to 3 years. The facial cutaneous abnormality, adenoma sebaceum, appears later in childhood, usually between the fourth and tenth years, and is progressive thereafter.
As the years pass, the seizures change pattern. In the first year or two they take the form of massive flexion spasms with hypsarrhythmia (irregular dysrhythmic bursts of high-voltage spikes and slow waves in the EEG). As many as 25 percent of patients with these types of seizures have been found to have tuberous sclerosis. Later, the seizures change to more typical generalized motor and psychomotor attacks or atypical petit mal. Any one of the seizure types may be brief, especially if the patient is receiving anti-epileptic medication. Focal neurologic abnormalities, which one might expect to occur from the size and location of some of the lesions, are distinctly uncommon.
Mental function continues to deteriorate slowly. Exceptionally there is a spastic weakness or mild choreoathetosis of the limbs and in a few cases an obstructive hydrocephalus develops. As in any state of severe developmental delay, a variety of nonspecific motor peculiarities—such as constant crying, muttering, stereotypical rocking and swaying movements, and digital mannerisms—may be observed. In nearly half of the cases, affective and behavioral derangements, often of hyperkinetic and aggressive type, are added to the intellectual deficiency.
The lack of parallelism in the severity of the epilepsy, the mental deficit, and cutaneous abnormalities has been noted by all clinicians who have wide experience with this disease. Some patients are subject to recurrent seizures while retaining relatively normal mental function; in others, trivial skin lesions or a retinal phakoma (see below) may suggest the diagnosis in a mentally normal person with few seizures. In such cases, recognition may elude competent neurologists and dermatologists. As a general rule, early onset of seizures is predictive of developmental delay. Gomez and colleagues suggested that the seizures damage the brain, a point with which we tend to agree in part. However, it seems likely that both the epilepsy and developmental delay are the product of severe involvement of the brain by the lesions of tuberous sclerosis.
Limitation of space allows no more than a catalogue of the other visceral abnormalities in tuberous sclerosis. In about half the cases, gray or yellow plaques (in reality gliomatous tumors) may be found in the retina in or near the optic disc or at a distance from it. It is from this lesion, called a phakoma, that van der Hoeve derived the term that is applied to all neurocutaneous diseases of this class. About half of all benign rhabdomyomas of the heart are associated with tuberous sclerosis; if located in the wall of the atrium, they may cause conduction defects. Other benign tumors of mixed cell type (angiomyolipomas) have been found in the kidneys, liver, lungs, thyroid, testes, and gastrointestinal tract. Cysts of the pleura or lungs, bone cysts in digits, and zones of marbling or densification in bones are some of the less common abnormalities.
Related posts:
 Disorders of the Special Senses
Chapter 20. Delirium and Other Acute Confusional States
Chapter 24. Fatigue, Asthenia, Anxiety, and Depression
Disorders of the Special Senses
Chapter 20. Delirium and Other Acute Confusional States
Chapter 24. Fatigue, Asthenia, Anxiety, and Depression
 Chapter 28. Normal Development and Deviations in Development of the Nervous System
Chapter 50. The Myotonias, Periodic Paralyses, Cramps, Spasms, and States of Persistent Muscle Fiber Activity
Chapter 28. Normal Development and Deviations in Development of the Nervous System
Chapter 50. The Myotonias, Periodic Paralyses, Cramps, Spasms, and States of Persistent Muscle Fiber Activity
 Chapter 48. Diseases of Muscle
Chapter 48. Diseases of Muscle
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


